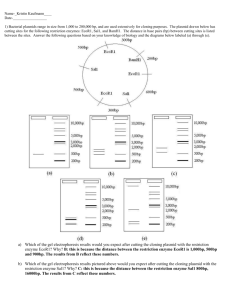
DOC
advertisement

1
Selecting Restriction Enzymes for Terminal Restriction Fragment Length
Polymorphism based on Phylogenetic Distance
K. J. Mei1 and S. H. Chang2
National Cheng Kung University, mq2kr3j92@hotmail.com
National Cheng Kung University, waterman2000.tw@gmail.com
1
2
ABSTRACT
An algorithm is proposed for choosing the most identical vector from a set of sample
vectors to match the given clusters. An application based on the algorithm for
choosing restriction enzymes according to the clusters has been developed.
Keywords: pattern identity, T-RFLP
1
INTRODUCTION
Terminal restriction enzyme fragment length polymorphism (T-RFLP) analysis is one of the most
common techniques for analyzing bacterial community (Liu, W. T. et al. 1997). This analysis simply
works through two facts: 1) different bacteria usually have different DNA codes 2) restriction enzyme
can recognize identical DNA code and cuts at those positions with identical DNA code. Briefly, DNA
fragments are generated from bacteria experimentally; and these DNA fragments are tagged
fluorescently at one end. Finally, restriction enzyme is applied to cut DNA fragments; only fragment
end is fluorescent, hence only these terminal fragments are detectable. Since different bacteria have
different DNA codes, restriction enzyme will cut at identical code yet different position, thus
generating terminal fragments with different lengths for different bacteria. For e.g. bacteria A has DNA
code “GGCCGATCGCCCT”, bacteria B has DNA code “GGCGGATCGGCCA”; if restriction enzyme that
cuts at middle “GGCC” is applied, it will generate “GG” for bacteria A and “GGCGGATCGG” for bacteria
B, and thus these two bacteria are distinguishable; however, if restriction enzyme that cuts at middle
“GATC” is applied, though it will generate different fragments “GGCCGA” and “GGCGGA”, their lengths
are the same and thus are not distinguishable.
This main problem always exists. Phylogenetic distantly-related bacteria, i.e. bacteria that are more
different based on evolution, sometimes share the same terminal restriction fragment length (T-RFL),
while phylogenetic closely-related bacteria sometimes have different T-RFLs. This may confuse
researchers since they may observe only one signal for several different bacteria, otherwise, many
signals for more similar bacteria (Engebretson, J. J. and Moyer, C. L. 2003).
Restriction enzyme is, therefore, one of the key factors towards successful T-RFLP analysis (Kitts,
C. L. 2001; Dickie, I. A. and FitzJohn, R. G. 2007). Ideally, researchers look for restriction enzyme that
may generate most different lengths for all bacteria. However, a clear guideline is currently lacking.
Usually, researchers decide suitable restriction enzymes by referring previous researches (Wells, G. F.
et al. 2009), or by testing different restriction enzymes towards samples experimentally (Zhang, R. et
al. 2008), or by simulating towards bacteria DNA code database (Kent, A. D. et al. 2003; Alvarado, P.
and Manjon, J. L. 2009).
To build logical method in choosing suitable restriction enzyme, an algorithm is developed.
Briefly, we try to match bacteria cluster information and sample vectors of restriction enzymes. The
bacteria cluster information tells whether two bacteria are considered similar or different. This cluster
information could be achieved through user-defined or through conversion from bacteria phylogenetic
distance matrix (distance matrix for every pair of bacteria DNA code after clustering analysis). In sum,
if sample vector of restriction enzyme is more matched towards cluster information, it indicates that
2
restriction enzyme is approaching nearer to the ideal “same T-RFL for similar bacteria/same cluster,
different T-RFLs for different bacteria/different clusters”.
2
ALGORITHM
An algorithm is proposed for choosing the most identical vector from a set of sample vectors to match
the given cluster. A program based on the algorithm for choosing restriction enzymes according to
clusters has been developed. By given cluster information and sample vectors of restriction enzymes,
user gets score for each sample vector. Cluster information and sample vectors of restriction enzymes
are first transfer into matrixes for distinction respectively. The clusters can be achieved by Userdefined or contributed by phylogenetic distance matrix. The distinct matrix of clusters then compares
to each distinct matrix of restriction enzymes. The scores for each sample vector of restriction enzyme
can be approached by matching and weighting between distinct matrixes. The weighting can be
achieved according to phylogenetic distance matrix or artificial weight. As shown in Fig. 1.
Cluster
By Phylogenetic
By User-defined
Distance Matris
Sample Vectors
of Restriction
Enzyme
Distinct
Matrix
Distinct
Matrixes
Match and Weight
By Phylogenetic
Distance Matris
By Artificial
Weight
Score
Fig. 1: Overview of selecting restriction Enzymes.
2.1
Distinct Matrix Establishment for Clusters
The clusters can be established by user-defined or phylogenetic distance matrix. Phylogenetic distance
matrix is an nxn upper triangle matrix. Values in the matrix is all within 0~1. By giving a cutoff value,
which has the same range, values in phylogenetic distance matrix can be classified into two values,
SAME and DIFFERENT. If the value in phylogenetic distance matrix is higher than the cutoff, the
classified value is DIFFERENT; otherwise, the classified value is SAME. The nxn upper triangle boolean
matrix for distinction, can be build according to the cutoff. As shown in Fig.2.
3
Phylogenetic
Distance Matrix
(nxn upper triangle
matrix, value between
0~1)
Distance Cutoff
(value between 0~1)
Compare distance
cutoff for each
element
Distinct Matrix for Cluster
(nxn upper triangle boolean
matrix, value within {SAME,
DIFFERENT})
Fig. 2: Construction of distinct matrix for phylogenetic distance matrix.
The user-defined cluster can be expanded to an nxn upper triangle boolean matrix. The value at an
element is SAME while the indexes are in the same cluster; otherwise, the value is DIFFERENT. A
sample for building distinct matrix is shown as Fig. 3. There are five species. There are three userdefined clusters and two clusters clustered by phylogenetic distance matrix and 3% cutoff, built into
an nxn upper triangle boolean matrix.
Fig. 3: A sample for building distinct matrix for clusters.
2.2
Distinct Matrix Establishment for Sample vector of Restriction Enzymes
Sample vector of restriction enzyme is an integer vector of length n. Assume there are m sample
vectors of restriction enzyme. The values in a vector are T-RFLs. Sometimes one restriction enzyme is
not enough to match the clusters. So combination of restriction enzymes is necessary. Through the
permutation of sample vectors, there are m* sample vectors of combined restriction enzymes.
4
Through the property that the restriction enzyme only cut the genes by side, the combination can be
done by chosen the smallest value for each element between vectors. After combination, make
difference matrixes for each sample vectors by pair-wise differentiate within each sample vector. The
difference matrix is with dimension nxn and each element in matrix is integer, totally m* matrixes.
The matrix for distinction can be approached by comparing with T-RFLP cutoff for all elements in the
difference matrix. If the value in the difference matrix is higher than the cutoff, the value in distinct
matrix is DIFFERENT; otherwise, the value is SAME. The distinct matrixes of restriction enzymes are m*
nxn upper triangle Boolean matrixes. As shown in Fig. 4.
Sample Vectors
of Restriction Enzyme
(m vectors of length
n, all value is integer)
Sample Vectors
of Restriction Enzymes
(m* vectors of length n, all
value is integer)
Build difference
matrixes by pair-wise
differentiate within
each sample vector
Distinct Matrixes of
Restriction Enzymes
(m* upper triangle boolean
matrixes of dimension
nxn, all value is within
{SAME, DIFFERENT})
Permutation
Degree of
Combination
Difference Matrixes of
Restriction Enzymes (m* upper
triangle matrixes of dimension
nxn, all value is integer)
Compare with T-RFLP
cutoff for all elements
for all matrixes
T-RFLP Cutoff
(integer)
Fig. 4: Construction of distinct matrixes for restriction enzymes.
A sample for building distinct matrix for restriction enzyme is shown in Fig. 5. The sample is with
T-RFLP cutoff 0 bp and a restriction enzyme with five T-RFLs for five species. By pair-wise comparison,
the vector of T-RFLs can be transfer into difference matrix. With T-RFLP Cutoff, the difference matrix
can be transfer into a boolean matrix for distinction.
5
Fig. 5: A sample for building distinct matrix for restriction enzyme.
2.3
Match and Weight between Distinct Matrixes for Cluster and Restriction Enzyme
There are two ways in this paper to weight between distinct matrixes for cluster and restriction
enzyme: by phylogenetic distance matrix or by artificial weight matrix. The phylogenetic distance
matrix must be the same dimension for distinct matrix; it is dimension of nxn, float matrix and all
values within 0~1. The artificial weight matrix is a 2x2 float matrix.
The artificial weight matrix is shown as Tab. 1. While matching elements by user-defined weight
matrix for matrixes, if both values are SAME, the result score is as weight value; if both values do not
match, the result score is 0; if both values are DIFFERENT, the result score is 1. Distinct matrix for
clusters matches and weights by weight matrix for each distinct matrixes for restriction enzymes. The
result is a score matrix for each restriction enzyme. Finally score matrixes are achieved; they are m*
nxn upper triangle float matrixes. As shown in Fig. 6.a. The phylogenetic distance matrix effects while
the element between distinct matrixes is equivalent. The score matrix is made by phylogenetic
distance matrix dotting the matched matrix. As shown in Fig. 6.b.
Restriction Enzyme\Cluster
SAME
DIFFERENT
SAME
Weight
0
DIFFERENT
0
1
Tab. 1: Artificial weight table for matching distinct matrixes of cluster and restriction enzyme.
Distinct Matrix
for Clusters
Artificial
Weight Matrix
(2x2 float matrix)
Distinct Matrixes for
Restriction Enzymes
Match and weight
by weight matrix
Score Matrixes
(m* nxn upper triangle
float matrixes)
Distinct Matrix
for Clusters
Phylogenetic
Distance Matrix
Distinct Matrixes for
Restriction Enzymes
Match and dot
phylogenetic
distance matrix
Score Matrixes
(m* nxn upper triangle
float matrixes)
6
Fig. 6: Match and weight between distinct matrixes for clusters and restriction enzymes: (a) by artificial
weight matrix, (b) by phylogenetic distance matrix
The scores for all restriction enzymes can be achieved by summing all elements in each score
matrixes and dividing the sums by sum of perfectly matched matrix. After sorting the scores, the best
restriction enzymes can be found. As shown in Fig. 7.
Score Matrixes
Sum all elements in each matrix.
Divide sum by sum of perfectly
matched matrix to get score
Scores
Sort and find the best
restriction enzymes
according to highest scores
The Best
Restriction Enzymes
Fig. 7: Find the best restriction enzymes through the score matrixes.
An equivalent sample of match and weight for a restriction enzyme followed with Fig. 3 and Fig. 5
is shown in Fig 8. The matched matrix is build by signed S while both elements in distinct matrix for
cluster and restriction enzyme are SAME, signed D while they are both DIFFERENT and signed N while
they are not equivalent. Only S and D get weight. If weighting by phylogenetic distance matrix, S and D
are no difference; they get the same weight according to phylogenetic distance matrix. The artificial
weight here is set as 0.1. S get the score of given weight value; D get the score of value 1. Finally there
are four kinds of result: (1) both cluster and weight by phylognetic distance matrix, (2) cluster by userdefined cluster and weight by phylognetic distance matrix, (3) cluster by phylognetic distance matrix
and weight by artificial weight, (4) cluster by user-defined cluster and weight by artificial weight.
7
Fig. 8: A sample for getting score by match and weight for four different ways.
3
PROGRAM APPLICATION
A program for the above algorithm is achieved. The user interface is separate to four parts: Cluster
operation console, restriction enzymes operation console, weight method selection console and score
console. As shown in the above labels in Fig. 9.
Cluster operation console is shown in Fig. 9. User can upload a phylogenetic distance matrix, set
the corresponding cutoff value; or upload a file for user-defined cluster. Restriction enzymes
operation console is shown in Fig. 10. The console can upload T-RFL vectors of restriction enzymes in
a matrix form and set the cutoff value for T-RFL and degree of combination. It can export combination
result. Weight method selection console is shown in Fig. 11. User can select to use phylogenetic
distance matrix as weighting in accordance or to use artificial weight. The score console presents the
corresponding T-RFLs for the selected restriction enzyme. As shown in Fig. 12, the row labels are
species; the figure above shows the clusters, which the red dash lines put on the same horizontal line
implies they are the same clusters; the column label of below figure shows the T-RFL. The title beyond
is the name of selected restriction enzyme. The graphs here are not perfectly matched.
Fig. 9: Cluster operation console.
8
Fig. 10: Restriction enzymes operation console.
Fig. 11: Weight method selection console.
Fig. 12: Score console presents the corresponding T-RFLs for the selected restriction enzyme.
4
DISCUSSION
The algorithm is like to choose the best “mask” for sample vectors of restriction enzymes. The mask
can be chosen and weighted from clusters or user-defined clusters.
Since within cluster, the difference for “natural weight”, which is based on phylogenetic distance
matrix, is smaller between clusters. The distinctibility causes the most weight. User often interests in
9
difference between classes then difference within class. So the “natural weight” for identical element is
reasonable no matter for clusters which based on phylogenetic distance matrix or for user-defined
clusters.
The artificial weight matrix is proposed as Tab. 1; the weight value is put on the position for both
values are “SAME” and the weight value is usually set within 0~1. If the weight value is set to 0, it
means the distinctibility is the only concern. The result can distinct most of species but not clusters; it
is still very meaningful, since the user knows how species clustered. If user can identify species, there
is no problem for clustering. If the weight value set much higher and close to 1, the feature of distinct
matrix of restriction enzyme will close to the distinct matrix of clusters. So, user can set weight value
to 1 first; if the result is not good enough; user can try the smaller weight value. The difference is user
should do cluster after mask. In the artificial weight matrix, there is 0 for (Cluster, Restriction
Enzyme)=(SAME, DIFFERENT). If there is weight, the best restriction enzyme will be chosen like Fig. 13,
all the elements are distinguishable.
Fig. 13: An example for the corresponding T-RFLs of the best restriction enzyme for weighting at
(Cluster, Restriction Enzyme)=(SAME, DIFFERENT) in artificial weighting matrix.
5
CONCLUSION
The algorithm for choosing the most identical vector from a set of sample vectors to match the given
clusters can be applied for choosing restriction enzymes according to the clusters. The clusters can be
achieved by User-defined or contributed by phylogenetic distance matrix. The weighting can be
achieved according to phylogenetic distance matrix or artificial weight. The application can provide a
free environment to manipulate clusters and selecting method.
REFERENCES
Alvarado, P. and Manjon, J. L. (2009). "Selection of enzymes for terminal restriction fragment length
polymorphism analysis of fungal internally transcribed spacer sequences." Appl Environ
Microbiol 75(14): 4747-4752.
Dickie, I. A. and FitzJohn, R. G. (2007). "Using terminal restriction fragment length polymorphism (TRFLP) to identify mycorrhizal fungi: a methods review." Mycorrhiza 17(4): 259-270.
Engebretson, J. J. and Moyer, C. L. (2003). "Fidelity of select restriction endonucleases in determining
microbial diversity by terminal-restriction fragment length polymorphism." Appl Environ
Microbiol 69(8): 4823-4829.
Kent, A. D., Smith, D. J., Benson, B. J. and Triplett, E. W. (2003). "Web-based phylogenetic assignment
tool for analysis of terminal restriction fragment length polymorphism profiles of microbial
communities." Appl Environ Microbiol 69(11): 6768-6776.
Kitts, C. L. (2001). "Terminal restriction fragment patterns: a tool for comparing microbial communities
and assessing community dynamics." Curr Issues Intest Microbiol 2(1): 17-25.
10
Liu, W. T., Marsh, T. L., Cheng, H. and Forney, L. J. (1997). "Characterization of microbial diversity by
determining terminal restriction fragment length polymorphisms of genes encoding 16S
rRNA." Appl Environ Microbiol 63(11): 4516-4522.
Wells, G. F., Park, H. D., Yeung, C. H., Eggleston, B., Francis, C. A. and Criddle, C. S. (2009). "Ammoniaoxidizing communities in a highly aerated full-scale activated sludge bioreactor:
betaproteobacterial dynamics and low relative abundance of Crenarchaea." Environ Microbiol.
Zhang, R., Thiyagarajan, V. and Qian, P. Y. (2008). "Evaluation of terminal-restriction fragment length
polymorphism analysis in contrasting marine environments." FEMS Microbiol Ecol 65(1): 169178.