27C
advertisement

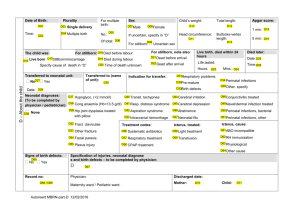
27C Perinatal Asphyxia Lisa M. Adcock Lu-Ann Papile I. PERINATAL ASPHYXIA refers to a condition of impaired gas exchange that leads, if persistent, to fetal hypoxemia and hypercarbia. It occurs during the first and second stage of labor and is identified by fetal acidosis, as measured in umbilical arterial blood. The umbilical artery pH that defines asphyxia of a sufficient degree to cause brain injury is unknown. Although the most widely accepted definition is a pH <7.0, even with this degree of acidosis the likelihood of brain injury is low. The following terms may be used in evaluating a term infant at risk for brain injury in the perinatal period: A. Neonatal depression is a general term used to describe an infant who has a prolonged transition from an intrauterine to an extrauterine environment. These infants usually have low 1- and 5minute Apgar scores. B. Neonatal encephalopathy is a clinical term used to describe an abnormal neurobehavioral state that consists of a decreased level of consciousness with abnormalities in neuromotor tone. It characteristically begins within the first postnatal day and may be associated with seizure-like activity, hypoventilation or apnea, depressed primitive reflexes and the appearance of brain stem reflexes. It does not imply a specific etiology, nor does it imply irreversible neurologic injury. C. Hypoxic-ischemic encephalopathy (HIE) is an abnormal neurobehavioral state in which the predominant pathogenic mechanism is impaired cerebral blood flow. D. Hypoxic-ischemic brain injury refers to neuropathology attributable to hypoxia and/or ischemia as evidenced by biochemical (such as serum creatine kinase brain bound [CK-BB]), electrophysiologic (EEG), neuroimaging (head ultrasonography [HUS], magnetic resonance imaging [MRI], computed tomography [CT]), or postmortem abnormalities. II. INCIDENCE. The frequency of perinatal asphyxia is approximately 1% to 1.5% of live births in the Western Hemisphere and is inversely related to gestational age and birth weight. It occurs in 0.5% of live born infants >36 weeks' gestation and accounts for 20% of perinatal deaths (50% if stillborns are included). A higher incidence is noted in term infants of diabetic or toxemic mothers, infants with intrauterine growth restriction, breech presentation, and postdates infants. III. ETIOLOGY. In term infants, 90% of asphyxial events occur in the antepartum or intrapartum period as a result of impaired gas exchange across the placenta that leads to the inadequate provision of oxygen (O2) and removal carbon dioxide (CO2) and H+ from the fetus. The remainder of these events occurs in the postpartum period and is usually secondary to pulmonary, cardiovascular, or neurologic abnormalities. A. Factors that increase the risk of perinatal asphyxia include the following: 1. Impairment of maternal oxygenation. 2. Decreased blood flow from mother to placenta. P.519 3. Decreased blood flow from placenta to fetus. 4. Impaired gas exchange across the placenta or at the fetal tissue level. 5. Increased fetal O2 requirement. B. Etiologies of perinatal hypoxia-ischemia include the following: 1. Maternal factors: hypertension (acute or chronic), infection, diabetes, hypotension, vascular disease, drug use, and hypoxia due to pulmonary, cardiac, or neurologic disease. 2. Placental factors: infarction, fibrosis, abruption, or hydrops. 3. Uterine rupture. 4. Umbilical cord accidents: prolapse, entanglement, true knot, compression. 5. Abnormalities of umbilical vessels. 6. Fetal factors: anemia, infection, cardiomyopathy, hydrops, severe cardiac/ circulatory insufficiency. 7. Neonatal factors: severe neonatal hypoxia due to cyanotic congenital heart disease, persistent pulmonary hypertension of the newborn (PPHN), cardiomyopathy, other forms of neonatal cardiogenic and/or septic shock. IV. PATHOPHYSIOLOGY A. Events that occur during the normal course of labor cause most babies to be born with little O2 reserve. These include the following: 1. Decreased blood flow to placenta due to uterine contractions, some degree of cord compression, maternal dehydration, maternal alkalosis due to hyperventilation. 2. Decreased O2 delivery to the fetus as a result of the reduction of placental blood flow. 3. Increased O2 consumption in both mother and fetus. B. During labor complicated by a hypoxic-ischemic challenge, the following changes may occur: 1. With brief asphyxia, there is a transient increase, followed by a decrease in heart rate (HR), mild elevation in blood pressure (BP), an increase in central venous pressure (CVP), and essentially no change in cardiac output (CO). This is accompanied by a redistribution of CO with an increased proportion going to the brain, heart and adrenal glands (diving reflex). 2. With prolonged asphyxia cerebral blood flow becomes dependent on systemic BP (loss of cerebral vascular autoregulation). A decrease in CO leads to hypotension and impaired cerebral blood flow resulting in anaerobic metabolism and eventual intracellular energy failure due to an increase in the utilization of glucose in the brain and a fall in the concentration of glycogen, phosphocreatine, and adenosine triphosphate (ATP). 3. Hypoxia-induced vascular dilatation increases glucose availability, at least transiently; and anaerobic metabolism produces lactic acid. C. Cellular changes occur due to diminished oxidative phosphorylation and ATP production. This energy failure impairs ion pump function, causing accumulation of intracellular Na+, Cl-, H2O, and Ca2+; extracellular K+; and excitatory amino acid (EAA) neurotransmitters (e.g., glutamate). Impairment of oxidative phosphorylation can occur during the primary asphyxial episode as well as during a secondary energy failure that usually occurs approximately 6 to 24 hours after the initiating insult. Cell death can be either immediate or delayed, and either apoptotic or necrotic. 1. Immediate neuronal death can occur due to intracellular osmotic overload of Na+ and Ca2+, as seen with excessive EAA acting on inotropic glutamate receptors (such as the N-methyl-D-aspartate [NMDA) receptor]) 2. Delayed neuronal death occurs secondary to uncontrolled activation of enzymes and second messenger systems within the cell (e.g., Ca2+-dependent lipases, proteases, and caspases); perturbation of mitochondrial respiratory electron chain transport; generation of free radicals and leukotrienes; generation of nitric oxide (NO) through NO synthase; or depletion of energy stores. 3. EAA also can activate α-3-hydroxy-5-methyl-isoxazole (AMPA) receptor channels, leading to oligodendrocyte progenitor cell death. P.520 4. Reperfusion of previously ischemic tissue may cause injury as it can promote the formation of excess reactive oxygen species (e.g., superoxide, hydrogen peroxide, hydroxyl, singlet oxygen), which can overwhelm the endogenous scavenger mechanisms, thereby causing damage to cellular lipids, proteins, and nucleic acids, as well as to the blood-brain barrier. This may result in an influx of neutrophils that, along with activated microglia, release injurious cytokines (e.g., interleukin 1-β [IL-1 β] and tumor necrosis factor α [TNF-α]). V. DIAGNOSIS A. Perinatal assessment of risk includes awareness of preexisting maternal or fetal problems that may predispose to perinatal asphyxia (see preceding list) and of changing placental and fetal conditions (see Chap. 1) ascertained by ultrasonographic examination, biophysical profile, nonstress tests, measurement of urinary estriol. B. Clinical presentation can be variable. Common clinical scenarios include a postdates infant with asphyxia, meconium aspiration, pulmonary hypertension, pneumothorax, or birth trauma. C. Low Apgar scores and need for resuscitation in the delivery room are common but nonspecific findings. Many features of the Apgar score relate to cardiovascular integrity and not neurologic function. 1. In addition to perinatal asphyxia, the differential diagnosis for a term infant with an Apgar score ≤3 for >5 minutes includes depression from maternal anesthesia or analgesia; trauma; metabolic or infectious insults; neuromuscular disorders; and central nervous system (CNS), cardiac, or pulmonary malformations 2. If the Apgar score is >6 by 5 minutes, perinatal asphyxia is not likely. D. Umbilical cord or first blood gas determination. The specific blood gas criteria that define asphyxia causing brain damage are uncertain. 1. In a population-based cohort of 17,000 term infants, the average umbilical cord arterial pH was 7.24 ± 0.07 and BE was -5.6 ± 0.3 mmol/L. Umbilical arterial pH <7.0 was present in only 0.4%. Of these, 5-minute Apgar score was <7 in 31% and <3 in 8.5%. The risk of adverse outcome was more likely if the acidosis is purely metabolic or mixed. 2. In another study, base deficit was measured in term infants who had persistent hemodynamic, respiratory, or neurologic abnormalities at 30 minutes of age. Metabolic acidosis with base deficit of 14 mmol/L or more had a sensitivity of 73.2% and a specificity of 82% in predicting moderate or severe neonatal encephalopathy. In two large randomized clinical trials of hypothermia for neonatal hypoxic/ischemic encephalopathy, severe acidosis was defined as pH of 7.0 or less or base deficit of ≥16 mmol/L. VI. HIE. The diagnosis of perinatal HIE requires an abnormal neurologic examination on the first day following birth. It is important to note that no significant neurologic abnormality diagnosed later in childhood (e.g., cerebral palsy [CP]) can be ascribed to perinatal asphyxia in the absence of evidence in the immediate neonatal period of neurologic abnormality and severe multiorgan dysfunction. A. The clinical spectrum of HIE is described as mild, moderate and severe (see Table 27C.1 Sarnat Stages of HIE). Infants can progress from mild to moderate and/or severe encephalopathy over the 72 hours following the hypoxic-ischemic insult. B. The diagnosis of neonatal encephalopathy includes a number of etiologies in addition to perinatal hypoxia-ischemia. Asphyxia may be suspected and HIE reasonably included in the differential diagnosis of term neonatal depression, coma, or neurologic dysfunction if the following have been documented: 1. Apgar score ≤3 at >5 minutes. 2. Fetal HR <60 beats/minute. 3. Prolonged (>1 hour) antenatal acidosis. 4. Seizures within first 24 to 48 hours after birth (50% of seizures are not asphyxial in etiology). 5. Burst-suppression pattern electroencephalography (EEG). P.521 TABLE 27C.1 Sarnat and Sarnat Stages of Hypoxic-Ischemic Encephalopathy* Stage Stage 1 (Mild) Stage 2 (Moderate) Stage 3 (Severe) Level of consciousness Hyperalert; irritable Lethargic or obtunded Stuporous, comatose Neuromuscular control: Uninhibited, Diminished spontaneous overreactive movement Diminished or absent spontaneous movement Muscle tone Normal Mild hypotonia Flaccid Posture Mild distal flexion Strong distal flexion Intermittent decerebration Stretch reflexes Overactive Overactive, disinhibited Decreased or absent Segmental myoclonus Present or absent Present Absent Complex reflexes: Normal Suppressed Absent Suck Weak Weak or absent Absent Moro Strong, low threshold Weak, incomplete high threshold Absent Oculovestibular Normal Overactive Weak or absent Tonic neck Slight Strong Absent Autonomic function: Generalized sympathetic Generalized parasympathetic Both systems depressed Pupils Mydriasis Miosis Midposition, often unequal; poor light reflex Respirations Spontaneous Spontaneous; occasional apnea Periodic; apnea Heart rate Tachycardia Bradycardia Variable Bronchial and salivary secretions Sparse Profuse Variable Gastrointestinal motility Normal or decreased Increased diarrhea Variable Seizures Common focal or Uncommon multifocal (6 to 24 hours of (excluding age) decerebration) None Electroencephalographic Normal findings (awake) Early: generalized lowEarly: periodic voltage, slowing pattern with (continuous delta and theta)isopotential phases Later: periodic pattern Later: totally (awake); seizures focal or isopotential multifocal; 1.0 to 1.5 Hz spike and wave Duration of symptoms <24 hours 2 to 14 days Hours to weeks Outcome About 100% 80% normal; abnormal if About 50% die; normal symptoms more than 5 to 7 remainder with days severe sequelae * The stages in this table are a continuum reflecting the spectrum of clinical states of infants over 36 weeks' gestational age. Source: From Sarnat H. B., Sarnat M. S. Neonatal encephalopathy following fetal distress: A clinical and electroencephalographics study. Arch Neurol 1976;33:696. P.522 6. Need for positive pressure ventilation for >1 minute or first cry delayed >5 minutes. VII. OTHER NEUROLOGIC CONSIDERATIONS A. Increased intracranial pressure (ICP), defined as >10 mm Hg, or cerebral edema should be regarded as an effect rather than a cause of brain damage. Cerebral edema peaks at 36 to 72 hrs after the insult. It often reflects extensive prior cerebral necrosis rather than swelling of intact cells, making this finding consistent with a uniformly poor prognosis. Efforts to reduce ICP and cerebral edema (high-dose phenobarbital, steroids, mannitol, and other hypertonic solutions) do not affect outcome. B. Seizures are described in 20% to 50% of infants with HIE, and usually start between 6 and 24 hours after the insult. They are most often seen in Sarnat stage 2 HIE, rarely in Sarnat stage 3, and almost never in Sarnat stage 1 HIE. 1. Seizures in HIE are usually subtle, tonic, or multifocal clonic. Generalized seizures are uncommon due to comparatively immature myelinization and synaptogenesis of the neonatal brain. Distinguishing between multifocal seizures and jitteriness (rhythmic segmental myoclonus) in stages 1 and 2 HIE may be difficult. They can be differentiated by holding the affected extremity and changing the tension on the muscle stretch receptor by slightly flexing or extending the joint. This should arrest clonus, whereas in true seizures, convulsive movements continue to be felt in the examiner's hand. 2. Seizures may be associated with increased cerebral metabolic rate, which could lead to further cerebral injury. 3. Seizures can compromise ventilation and oxygenation, especially in infants who are not on mechanical ventilation. In infants on musculoskeletal blockade for mechanical ventilation, seizures may be manifested by abrupt changes in BP, HR and oxygenation. 4. Seizures associated with HIE are often very difficult to control. Whether seizures alone, in the absence of metabolic or cardiopulmonary abnormalities, lead to brain injury is controversial. VIII. MULTIORGAN DYSFUNCTION. Other organ systems in addition to the brain usually exhibit evidence of asphyxial damage. A. In some cases, the brain may be the only organ exhibiting dysfunction following asphyxia. In one series of 57 infants, HIE occurred without other system involvement in 14 (24.5%). B. The gamut of organ involvement in perinatal asphyxia varies among series, depending in part upon the definitions used for asphyxia and organ dysfunction. 1. In a retrospective study of 130 term infants with asphyxia, the proportion of those with organ dysfunction was: renal 70%, cardiovascular 62%, pulmonary 86%, hepatic 85%. Infants were diagnosed with asphyxia if they needed mechanical ventilation at birth, exhibited encephalopathy, and had one or more of the following: (i) 5-minute Apgar score <5, (ii) Base deficit 16 or more mmol/L documented within first hour of life, and (iii) delayed respiratory effort for 5 or more minutes of life. 2. In another series of 152 asphyxiated term infants followed prospectively, neurologic and systemic complications occurred in 43% and 57%, respectively. Organ dysfunction included respiratory abnormalities 39%, infection 17%, gastrointestinal intolerance 15%. Infants were considered to have asphyxia if they had fetal distress, were depressed at birth, and exhibited a metabolic acidosis. C. Multiorgan dysfunction is theorized to be secondary to the “diving reflex” (see IV B 1). 1. The kidney is the most common organ to be affected in perinatal asphyxia. The proximal tubule of the kidney is especially affected by decreased perfusion, leading to acute tubular necrosis (ATN) (see Chap. 31). 2. Cardiac dysfunction is caused by transient myocardial ischemia. The ECG may show ST depression in the midprecordium and T-wave inversion in the left precordium. Echocardiographic findings include decreased left ventricular P.523 contractility, especially of posterior wall; elevated ventricular end-diastolic pressures; tricuspid insufficiency and pulmonary hypertension due to poor ventricular function. In severely asphyxiated infants, dysfunction more commonly affects the right ventricle. A fixed HR may raise suspicion of clinical brain death. 3. Gastrointestinal effects include an increased risk of bowel ischemia and necrotizing enterocolitis (see Chap. 32). 4. Hematologic effects include disseminated intravascular coagulation due to damage to blood vessels, poor production of clotting factors due to liver dysfunction, and poor production of platelets by the bone marrow. 5. Liver involvement may be manifested by isolated elevation of hepatocellular enzymes. More extensive damage may occur, leading to DIC, inadequate glycogen stores with resultant hypoglycemia, or altered detoxification or elimination of drugs. 6. Pulmonary effects include increased pulmonary vascular resistance leading to PPHN, pulmonary hemorrhage, pulmonary edema due to cardiac dysfunction, secondary RDS due to failure of surfactant production, and meconium aspiration. IX. LABORATORY EVALUATION OF EFFECTS OF ASPHYXIA A. Cardiac evaluation 1. Cardiac troponin I (cTNI) and cardiac troponin T (cTnT), cardiac regulatory proteins that control the calcium-mediated interaction of actin and myosin, are markers of myocardial damage. Normal values in the neonate are troponin I = 0 - 0.28 ± 0.42 µg/L and troponin T = 0 - 0.097 µg/L. Elevated levels of these proteins have been described in infants with clinical and laboratory evidence of asphyxia. 2. An elevation of serum creatine kinase myocardial bound (CK-MB) fraction of >5% to 10% may indicate myocardial injury. B. Brain injury. 1. Serum CK-BB. This may be increased in asphyxiated infants within 12 hours of the insult, but has not been correlated with long-term neurodevelopmental outcome. CK-BB is also expressed in placenta, lungs, gastrointestinal tract, and kidneys. 2. In one report, measurement of protein S-100 (>8.5 µg/L) plus elevated CK-BB, or elevated CK-BB and low cord blood arterial pH had sensitivity of 71% each and specificity of 95% and 91% respectively in predicting moderate to severe encephalopathy. C. Renal evaluation 1. Blood urea nitrogen (BUN) and serum creatinine (Cr) may be elevated in perinatal asphyxia. Typically elevation is noted 2 to 4 days after the insult. 2. Fractional excretion (FE) of Na+ or renal failure index may help confirm renal insult (see Chap. 31). 3. Urine levels of β-2-microglobulin have been used as an indicator of proximal tubular dysfunction, although not routinely. This low molecular weight protein is freely filtered through the glomerulus and reabsorbed almost completely in the proximal tubule. 4. Renal sonographic abnormalities correlate with the occurrence of oliguria. X. CRANIAL IMAGING A. Cranial sonographic examination is less useful than other imaging modalities in assessing edema, subtle midline shift, superficial cortical or posterior fossa hemorrhage, and ventricular compression. B. Computed tomography (CT) may be useful for determining the extent of cerebral edema, especially when performed 2 to 4 days after the insult. C. Magnetic resonance imaging (MRI). T1- and T2-weighted MRI has been considered the best modality for imaging the neonatal brain; however, standard MRI may not detect hyoxic-ischemic changes during the first few days after the insult. High signal on T2-weighted images represents vasogenic edema. 1. Diffusion-weighted images (DWI) can show abnormalities within hours of the insult that may yield prognostic information. By detecting differences in rates P.524 of diffusion of water protons, DWI reveals restricted water diffusion, reflecting cytotoxic edema that is not apparent on conventional MRI. However, DWI does not distinguish cytotoxic edema from cell death, especially in global diffuse injuries, during the first hours following a hypoxic-ischemic insult. 2. Localized magnetic resonance spectroscopy (MRS), also called proton-MRS or 1HMRS, measures the relative concentrations of various metabolites in tissue. Elevated lactate and abnormal ratios of choline to total creatine and N-acetylaspartate (NAA) to total creatine have been described following neonatal hypoxic-ischemic brain injury and may yield prognostic information. XI. EEG is used both to evaluate for seizure activity and also to define abnormal background activity such as burst-suppression, continuous low voltage, or isoelectric patterns. When expertise in interpretation of neonatal EEGs is not readily available, amplitude integrated EEG (aEEG) has been used to evaluate for seizures and to define abnormal background patterns. This method consists of a single-channel EEG from biparietal electrodes. There is selective filtering of specific channels (<2 Hz and >15 Hz), then integration of the signal amplitude and semilogarithmic recording of the processed signal. XII. PATHOLOGIC FINDINGS OF BRAIN INJURY A. Specific neuropathology may be seen after moderate or severe asphyxia. 1. Focal or multifocal cortical necrosis affecting all cellular elements can result in cystic encephalomalacia and/or ulegyria (attenuation of depths of sulci) due to loss of perfusion in one or several vascular beds. 2. Watershed infarcts occur in boundary zones between cerebral arteries, particularly following severe hypotension. They reflect poor perfusion of the vulnerable periventricular border zones in the centrum semiovale and produce predominantly white matter injury. In the term infant, this typically results in bilateral parasagittal cortical and subcortical white matter injury or injury to the parieto-occipital cortex. 3. Selective neuronal necrosis is the most common type of injury seen following perinatal asphyxia. It is due to differential vulnerability of specific cell types; for example, neurons are more easily injured than glia. Specific regions at increased risk are CA1 region of hippocampus, Purkinje cells of cerebellum in term infants, and brainstem nuclei. Necrosis of thalamic nuclei and basal ganglia (status marmoratus) can be considered a subtype of selective neuronal necrosis. B. Neuropathology may reflect the type of asphyxial episode, although the precise pattern is not predictable. 1. Prolonged partial episodes of asphyxia tend to cause diffuse cerebral (especially cortical) necrosis. Expected clinical findings usually include seizures and paresis. 2. Acute total asphyxia tends to spare the cortex although affecting primarily the brainstem, thalamus, and basal ganglia. Expected clinical findings usually include disturbances in consciousness, respiration, HR, BP, and temperature control; disorders of tone and reflexes; cranial nerve palsies. 3. Partial prolonged asphyxia followed by a terminal acute asphyxial event (combination) is probably present in most cases. XIII. TREATMENT A. Perinatal management of high-risk pregnancies 1. Fetal HR and rhythm abnormalities may provide supporting evidence of asphyxia, especially if accompanied by presence of thick meconium. However, they provide no information concerning duration or severity of an asphyxial event. 2. Measurement of fetal scalp pH is a better determinant of fetal oxygenation than Po2. With intermittent hypoxia-ischemia, Po2 may improve transiently whereas the pH progressively falls. Fetal scalp blood lactate has been suggested as easier and more reliable than pH, but has not gained wide acceptance. 3. Close monitoring of progress of labor with awareness of other signs of in utero stress. P.525 4. The presence of a constellation of abnormal findings may indicate the need to mobilize the perinatal team for a newborn that could require immediate intervention. Alteration of delivery plans may be indicated and guidelines for intervention in cases of suspected fetal distress should be designed and in place in each medical center (see Chap. 1). B. Delivery room management (see Chaps. 4, 17, and 24). The initial management of the hypoxic-ischemic infant in the delivery room is described in Chapter 4. C. Postnatal management of neurologic effects of asphyxia 1. Ventilation. CO2 should be maintained in the normal range. Hypercapnia can cause cerebral acidosis and cerebral vasodilation. This may result in more flow to uninjured areas and relative ischemia to damaged areas (“steal phenomenon”). Excessive hypocapnia (CO2 <25 mm Hg) may decrease CBF. 2. Oxygenation. Oxygen levels should be maintained in the normal range, although poor peripheral perfusion may limit the accuracy of continuous noninvasive monitoring. Hypoxemia should be treated with supplemental O2 and/or ventilation. Hyperoxia may cause decreased CBF or exacerbate free radical damage. 3. Temperature should be maintained in the normal range and hyperthermia should be avoided. 4. Perfusion. Cardiovascular stability and adequate mean systemic arterial BP are important in order to maintain adequate cerebral perfusion pressure. 5. Maintain physiologic metabolic state a. Hypocalcemia is a common metabolic alteration after neonatal asphyxia. It is important to maintain calcium in the normal range, because hypocalcemia can compromise cardiac contractility and may cause seizures (see Chap. 29B, Hypocalcemia, Hypercalcemia, and Hypermagnesemia Glucose); (see Chaps. 27A). b. Hypoglycemia is often seen in asphyxiated infants. Blood glucose level should be maintained in the normal range for term infants. Hyperglycemia may lead to an increase in brain lactate, damage to cellular integrity, increased edema, or further disturbance in vascular autoregulation. Hypoglycemia may potentiate excitotoxic amino acids. 6. Judicious fluid management is needed and fluid overload should be avoided. Two processes predispose to fluid overload in asphyxiated infants: a. Syndrome of inappropriate secretion of antidiuretic hormone (SIADH) (see Chap. 9) is often seen 3 to 4 days after the hypoxic-ischemic event. It is manifested by hyponatremia and hypo-osmolarity in combination with inappropriately concentrated urine (elevated urine specific gravity, osmolarity, and Na+). b. ATN (see Chap. 31) can result from the “diving reflex” (see preceding text). c. Fluid restriction may aid in minimizing cerebral edema although the effect of fluid restriction on long-term outcome in infants who are not in renal failure is not known. 7. Control of seizures. Seizures secondary to asphyxia are generally self-limited to the first few postnatal days. Because they are extremely difficult to control, it may not be possible to eliminate them completely. Once levels of conventional anticonvulsants are maximized, there is little utility in eliminating every “twitch” or electrographic seizure unless there is cardiopulmonary compromise from the seizures. In infants on musculoskeletal blockade, seizures may be manifested by abrupt changes in BP, HR, and oxygenation. Whether seizures, per se, cause brain injury is unknown. There is inadequate evidence to support the continued use of anticonvulsants in the absence of clinical or electrical (EEG) seizures. Metabolic perturbations such as hypoglycemia, hypocalcemia, hyponatremia, should be excluded before initiating anticonvulsant therapy. P.526 a. Acute use of anticonvulsants i. Phenobarbital is the initial drug of choice. It is given as a loading dose of 20 mg/kg IV. If seizures continue, an additional loading dose of 10 to 20 mg/kg IV may be given. A maintenance dose of 3 to 5 mg/kg/day PO or IV divided BID should be started 12 to 24 hours after the loading dose. Intramuscular (IM) is avoided because absorption is too slow. During initiation, the infant needs to be monitored closely for respiratory depression. Therapeutic serum levels are 20 to 40 mg/dL. Because a prolonged serum half-life due to renal compromise may result in drug accumulation, serum levels need to be monitored closely and the maintenance dose adjusted accordingly. ii. Phenytoin is usually added when seizures are not controlled by phenobarbital. The loading dose is 15 to 20 mg/kg IV followed by a maintenance dose of 4 to 8 mg/kg/day. In many centers, phosphenytoin is used in place of parent drug (phenytoin) because the risk of hypotension is less and extravasation has no adverse effects. Dosage is calculated and written in terms of phenytoin equivalents to avoid medication errors. Therapeutic serum level is typically 20 mg/dL. iii. Benzodiazapenes are considered third-line drugs and include lorazepam 0.05 to 0.1 mg/kg/dose IV. b. Long-term anticonvulsant management. Anticonvulsant therapy can be weaned when the clinical exam and EEG indicate that the infant is no longer having seizures. If the infant is receiving more than one anticonvulsant, weaning should be in the reverse order of initiation, with phenobarbital being weaned last. Phenobarbital is then tapered over several weeks. If there is EEG evidence of seizure activity, phenobarbital should be continued for 3 to 6 months. Approximately 25% of infants will need ongoing anticonvulsant therapy. Infants who have a high risk of recurring seizures in infancy or childhood are those with persistent neurologic deficit (50%) and those with an abnormal EEG between seizures (40%) 8. Management of other target organ injury a. Cardiac dysfunction should be managed with correction of hypoxemia, acidosis, and hypoglycemia and avoidance of volume overload. Diuretics may not be helpful if concomitant renal impairment is present. Infants will require continuous monitoring of systemic mean arterial BP, CVP (if available), and urine output. Infants with cardiovascular compromise may require inotropic drugs such as dopamine (see Chap. 17) and may need afterload reduction with a peripheral β-antagonist (e.g., isoproterenol) or phosphodiesterase inhibitor (e.g., milrinone) to maintain BP and perfusion. i. Arterial BP should be maintained in the normal range to support adequate cerebral perfusion. ii. Monitoring of CVP may be helpful to assess adequacy of preload (i.e., that the infant is not hypovolemic due to vasodilatation or third spacing); a reasonable goal is 5 to 8 mm Hg in term infants. b. Renal dysfunction should be monitored by measuring urine output, and with urinalysis, urine specific gravity, paired urine/serum osmolarity and serum electrolytes. i. In the presence of oliguria or anuria avoid fluid overload by limiting free water administration to replacement of insensible losses and urine output (~60 mL/kg/day) and consider using low-dose dopamine infusion (≤5 µg/kg/min) (see Chaps. 9 and 31). ii. Volume status should be evaluated before instituting strict fluid restriction. If there is no or low urine output, a 10 to 20 mL/kg fluid challenge followed by a loop diuretic such as furosemide may be helpful. iii. To avoid fluid overload, as well as hypoglycemia, concentrated glucose infusions delivered through a central line may be needed. Glucose levels P.527 should be monitored closely and rapid glucose boluses avoided. Infusions should be weaned slowly to avoid rebound hypoglycemia. c. Gastrointestinal effects. Feeding should be withheld until good bowel sounds are heard and stools are negative for blood and/or reducing substances (see Chap. 32). d. Hematologic abnormalities (see Chap. 26). Coagulation profile is monitored with partial thromboplastin time (PTT) and prothrombin time (PT), fibrinogen, and platelets. Abnormalities may need to be corrected with fresh frozen plasma, cryoprecipitate, and/or platelet infusions. e. Liver function should be monitored with measurement of transaminases (ALT, AST), clotting (PT, PTT, fibrinogen), albumin, bilirubin, and ammonia. Levels of drugs that are metabolized or eliminated through the liver must be monitored. f. Lung (see Chap. 24). Management of the pulmonary effects of asphyxia depends on the specific condition. XIV. NEUROPROTECTIVE STRATEGIES. A number of neuroprotective strategies have been proposed. A. Agents tested in animals with little data in human newborns include antagonists of excitotoxic neurotransmitter receptors such as NMDA receptor blockade with ketamine or MK-801; free radical scavengers such as allopurinol, superoxide dismutase, and vitamin E; Ca2+-channel blockers such as magnesium sulfate, nimodipine, nicardipine; cyclooxygenase inhibitors such as indomethacin; benzodiazepine receptor stimulation such as midazolam; and enhancers of protein synthesis such as dexamethasone. B. Mild induced hypothermia used under strict experimental protocols may be a potentially useful treatment for acute perinatal asphyxia based on short-term outcomes (18 months) in two randomized clinical trials. However, the results of ongoing trials and long-term efficacy and safety need to be established before this therapeutic modality can be considered standard of care. XV. OUTCOME IN PERINATAL ASPHYXIA A. The overall mortality rate is 10% to 30%. The frequency of neurodevelopmental sequelae in surviving infants is approximately 15% to 45%. B. The risk of CP in survivors of perinatal asphyxia is 5% to 10% compared to 0.2% in the general population. Most CP is not related to perinatal asphyxia, and most perinatal asphyxia does not cause CP. Only 3% to 13% of infants with CP have evidence of intrapartum asphyxia. C. Specific outcomes depend on the severity of the encephalopathy, the presence or absence of seizures, EEG results, and neuroimaging findings 1. Severity of encephalopathy can be ascertained using the Sarnat clinical stages of HIE (Table 27C.1). a. Stage 1 HIE: 98% to 100% of infants will have a normal neurologic outcome and < 1% mortality. b. Stage 2 HIE: 20% to 37% die or have abnormal neurodevelopmental outcomes. Infants who exhibit Stage 2 signs for >7 days have poorer outcomes. In one study, half of the 42 surviving infants who had Sarnat stage 2 encephalopathy had normal neurodevelopment at 1 year of age; approximately 10% had a normal neurologic exam and mild developmental delay and one-third were diagnosed with CP. c. Stage 3 HIE: 50% to 89% die and all survivors have major neurodevelopmental impairment. d. Prognosis is considered to be good if an infant does not progress to and/or remains in stage 3 and if total duration of stage 2 is <5 days. e. Some neurologically normal survivors of perinatal asphyxia have problems in school. In one study, all stage 1 HIE and 65% to 82% of stage 2 HIE children performed at expected grade level at 8 years. In another study, children 8 to 13 years' old who had neonatal encephalopathy plus Apgar score <4 had increased risk of problems with mathematics (3.3 times higher), problems with reading (4.6 times higher), epilepsy (7 times higher), P.528 minor motor problems (13 times greater), attention deficit-hyperactivity disorder (14 times greater) compared to unaffected children. 2. The presence of seizures increases an infant's risk of CP 50 to 70 fold. Mortality risk is highest for seizures that begin within 12 hours of birth (53%). In one study, infants whose seizure duration was 1 day had a 7% rate of CP and 11% had epilepsy on followup. If seizures lasted for >3 days, the rates of CP and epilepsy were 46% and 40% respectively. 3. The detection of low voltage activity, electrocerebral inactivity or burst-suppression patterns on EEG is a better prognostic indicator of poor outcome than is the finding of epileptiform activity. In particular, 93% of neonates with extreme burst suppression activity have poor outcomes. Persistent burst suppression is associated with an 86% to 100% risk of death or severe neurodevelopmental sequelae. 4. Normal findings on DWI MRI between 2 and 18 days of age are associated with normal neuromotor outcome at 12 to 18 months. Abnormalities of deep gray matter that are detected early have the worse motor and cognitive outcomes. In one study, abnormal DWI of the basal ganglia noted within 10 days of a hypoxic-ischemic insult was associated with a 93% risk of abnormal neurodevelopmental outcome at 9 months to 5 years. Suggested Readings ACOG Task Force on Neonatal Encephalopathy and Cerebral Palsy. Neonatal encephalopathy and cerebral palsy: Defining the pathogenesis and pathophysiology. Washington, DC: American College of Obstetricians and Gynecologists, 2003. Blackmon LR, Stark AR. Hypothermia: A neuroprotective therapy for neonatal hypoxic-ischemic encephalopathy. Pediatrics 2006;117:942-948. Edwards AD, Azzopardi DV. Therapeutic hypothermia following perinatal asphyxia. Arch Dis Child Fetal Neonatal Ed 2006;91:F127-F131. Higgins RD, Raju TNK, Perlman J, et al. Hypothermia and perinatal asphyxia: Executive summary of the National Institute of Child Health and Human Development Workshop. J Pediatr 2006;148:170-175.