Supplementary Information (doc 410K)
advertisement
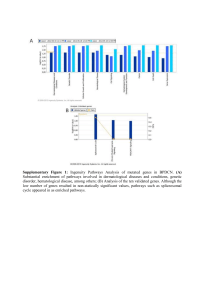
")
METHODS Patient samples DNA samples for whole exome sequencing (WES) were obtained from normal tissue (oral mucosa) and tumor bone marrow. The study was approved by the ethical research and animal care committee from the Institute of Health Carlos III (CEI PI 32_2009). DNA was extracted by the phenol–chloroform method and the quality and quantity of purified DNA was assessed by fluorometry (Qubit, Invitrogen) and gel electrophoresis. WES and bioinformatic analysis The preparation of shotgun libraries from the leukemic and non-leukemic genomic DNA followed the “SureSelect Human All Exon Kit” protocol (Agilent Technologies, Santa Clara, CA), covering 50 Mb of coding exons (approximately 1.60% of the genome). One to three µg of high molecular weight genomic DNA from each sample was fragmented by acoustic shearing on a Covaris S2 instrument. Fractions of 150–300 bp were ligated to Illumina adapters and PCR-amplified for 6 cycles. For exon enrichment, 300 ng of library were hybridized to SureSelect Human All Exon Capture kit for 24 h at 65ºC. Biotinylated hybrids were captured, and the enriched libraries were completed by amplifying with 12 cycles of PCR. The resulting purified DNA library was applied to an Illumina flow cell for cluster generation and sequenced using the Illumina Genome Analyzer IIx generating 2x76 base pair, paired-end reads by following the manufacturer protocols. Exome variant analysis and biological impact predictions were performed by RUbioSeq software [1] using default parameters for somatic variation analysis. In detail, sequencing data were first checked by FastQC for quality control checks on raw sequence data and then aligned to the human reference genome (GRCh37) using Burrows-Wheeler alignment (BWA) [2]. That reads unmapped by BWA were realigned using BFAST [3]. Somatic variants were identified using the Unified Genotyper v2 available at the GATK [4]. For variant calling we used GATK Unified Genotyper v2 applying the ‘‘Discovery’’ genotyping mode and default parameters for filtering. Thus, SNPs available at dbSNP 132 (hg19) and those reported by the 1000 Genomes Project, as well as changes present in the matched normal DNA and silent changes, were filtered out from VCF output files [5]. The GATK QUAL field was employed for ranking selected somatic variants. Biological impact predictions for detected variants were obtained from Ensembl Variant Effect Predictor [6]. In order to estimate the damaging character of the missense mutations, we used two different algorithms: SIFT and PolyPhen-2. A workflow of WES and the bioinformatic analysis is provide in Supplementary Figure 2. We used polymerase-chain-reaction (PCR) amplification and direct DNA sequencing to validate candidate variants. Because the sensitivity of Sanger, sequence variants that were reported in less than 20% of the reads could not be included in this validation phase. PCR products were cleaned using ExoSAP-IT (USB) and sequencing using ABI Prism BigDye terminator v3.1 (Applied Biosystems) with 5pmol of each primer. Sequencing reactions were run on an ABI-3730 automated sequencer (Applied Biosystems). All sequences were manually examined. IKZF3, ASXL1 and TP53 screening PCR and Sanger sequencing of IKZF3, ASXL1 and TP53 were performed in 26 additional CML samples (13 CP and 13 BC/NCgR). Primers spanning all coding exons of IKZF3, exon 13 of ASXL1 and all coding exons of TP53 are reported in Supplementary Table 3. PCR products were cleaned using ExoSAP-IT (USB) and sequencing using ABI Prism BigDye terminator v3.1 (Applied Biosystems) with 5pmol of each primer. Sequencing reactions were run on an ABI-3730 automated sequencer (Applied Biosystems). All sequences were manually examined. Supplementary Table 1: SNSs filtering steps of CML WES Filter CP CCyR BC Total 3123 7678 3306 Somatic 719 1839 869 Exonic 372 878 412 Consequence* 77 81 66 Quality Control 13 7 15 *Frameshift, stop gain and non-synonymous-deleterious CML-Phase Position CP 5_33549323_G/T CP 5_5232589_G/A CP 20_31022550_G/T CP 1_207305072_C/G CP 4_1388623_-/CA CP 2_71801335_-/AGGCGG CP 17_37922621_C/T CP 4_23825925_23825926delinsCT CP 1_25824890_A/G CP 17_7577551_C/T CP 15_41870343_C/T CP 21_46197270_T/A CP 2_145157495_A/C CCyR 4_1388623_-/CA CCyR 12_53207608_-/CAG CCyR 22_29885623_-/AGGAAG CCyR 5_43613188_TGCCCAAATCCAA/CCyR 13_25671311_TATGA/CCyR 13_21729832_-/TGGAATTT CCyR 17_7577551_C/T BC 1_12785494_G/BC 5_33549323_G/T BC 5_5232589_G/A BC X_2835999_CCACGCCGG/BC 20_31022550_G/T BC 1_207305072_C/G BC 4_1388623_-/CA BC 3_75714825_G/BC 17_37922621_C/T BC 4_23825925_23825926delinsCT BC 1_25824890_A/G BC 17_7577551_C/T BC 15_41870343_C/T BC 21_46197270_T/A BC 2_145157495_A/C Gene Name ADAMTS12 ADAMTS16 ASXL1 C4BPA CRIPAK DYSF IKZF3 PPARGC1A TMEM57 TP53 TYRO3 UBE2G2 ZEB2 CRIPAK KRT4 NEFH NNT PABPC3 SKA3 TP53 AADACL3 ADAMTS12 ADAMTS16 ARSD ASXL1 C4BPA CRIPAK FRG2C IKZF3 PPARGC1A TMEM57 TP53 TYRO3 UBE2G2 ZEB2 QUAL Coverage Freq_Var(%) Consequence 503,94 24 8 NON_SYNONYMOUS_CODING 1206,77 96 50 NON_SYNONYMOUS_CODING 323,4 27 48 STOP_GAINED 1060,02 57 61 STOP_GAINED 488,27 122 9 FRAMESHIFT_CODING 320,44 24 20 FRAMESHIFT_CODING 707,81 50 48 NON_SYNONYMOUS_CODING 999,13 113 35 NON_SYNONYMOUS_CODING 2076,54 155 50 NON_SYNONYMOUS_CODING 768,45 47 62 NON_SYNONYMOUS_CODING 244,25 23 4 NON_SYNONYMOUS_CODING 1222,13 87 52 NON_SYNONYMOUS_CODING 954,8 67 55 NON_SYNONYMOUS_CODING 427,91 141 9 FRAMESHIFT_CODING 1437,89 198 5 IN-FRAME 894,64 248 4 IN-FRAME 264,02 91 18 FRAMESHIFT_CODING 225,27 197 9 FRAMESHIFT_CODING 1464,5 81 9 SPLICE_SITE,FRAMESHIFT_CODING 250,69 45 27 NON_SYNONYMOUS_CODING 575,54 187 5 FRAMESHIFT_CODING 633,95 46 50 NON_SYNONYMOUS_CODING 723,43 103 30 NON_SYNONYMOUS_CODING 278,35 29 17 IN-FRAME 390,7 35 49 STOP_GAINED 511,16 53 36 STOP_GAINED 501,39 95 14 FRAMESHIFT_CODING 294,25 243 20 FRAMESHIFT_CODING 618,13 57 39 NON_SYNONYMOUS_CODING 1318,91 117 41 NON_SYNONYMOUS_CODING 1452,52 199 29 NON_SYNONYMOUS_CODING 228,95 17 50 NON_SYNONYMOUS_CODING 142,13 38 20 NON_SYNONYMOUS_CODING 1439,23 104 50 NON_SYNONYMOUS_CODING 808,39 69 45 NON_SYNONYMOUS_CODING DNA Level (cDNA) NM_030955.2:c.4291C>A NM_139056.2:c.1810G>A NM_015338.5:c.2035G>T NM_000715.3:c.1071C>G NM_175918.3:c.324_325insCA NM_001130987.1:c.3236delinsAGGCGG NM_012481.3:c.952G>A NM_013261.3:c.854_855delinsAG NM_018202.4:c.1928A>G NM_000546.4:c.730G>A NM_006293.3:c.2542C>T NM_003343.5:c.188A>T NM_014795.3:c.1259T>G NM_175918.3:c.324_325insCA NM_002272.2:c.457_458insCAG NM_021076.3:c.1994_1995insAGGAAG NM_012343.3:c.330_342del NM_030979.2:c.976_980del NM_145061.5:c.1238_1238+1insTGGAATTT NM_000546.4:c.730G>A NM_001103170.1:c.584del NM_030955.2:c.4291C>A NM_139056.2:c.1810G>A NM_001669.2:c.701_709del NM_015338.5:c.2035G>T NM_000715.3:c.1071C>G NM_175918.3:c.324_325insCA NM_001124759.1:c.483del NM_012481.3:c.952G>A NM_013261.3:c.854_855delinsAG NM_018202.4:c.1928A>G NM_000546.4:c.730G>A NM_006293.3:c.2542C>T NM_003343.5:c.188A>T NM_014795.3:c.1259T>G Protein Level p.Pro1431Thr p.Gly604Arg p.Gly679* p.Tyr357* p.Thr109Glnfs*82 p.Glu1081Glyfs*59 p.Glu318Lys p.Ser285Lys p.Lys643Arg p.Gly244Ser p.Arg848Trp p.Asp63Val p.Leu420Arg p.Thr109Glnfs*82 p.Gly152_Gly153insAla p.Lys665_Ala666insGlyArg p.Ala111Glyfs*23 p.Met326Glyfs*21 p.*413Cysfs*4 p.Gly244Ser p.Cys195Phefs*17 p.Pro1431Thr p.Gly604Arg p.Ala234_Val236del p.Gly679* p.Tyr357* p.Thr109Glnfs*82 p.Arg161Serfs*5 p.Glu318Lys p.Ser285Lys p.Lys643Arg p.Gly244Ser p.Arg848Trp p.Asp63Val p.Leu420Arg PolyPhen probably_damaging probably_damaging NA NA NA NA probably_damaging probably_damaging probably_damaging probably_damaging probably_damaging probably_damaging probably_damaging NA NA NA NA NA NA probably_damaging NA possibly_damagin possibly_damagin NA NA NA NA NA possibly_damagin possibly_damagin possibly_damagin possibly_damagin possibly_damagin possibly_damagin possibly_damagin SIFT tolerated deleterious NA NA NA NA deleterious deleterious tolerated deleterious deleterious deleterious deleterious NA NA NA NA NA NA deleterious NA tolerated deleterious NA NA NA NA NA deleterious deleterious tolerated deleterious deleterious deleterious deleterious Supplementary Table 2: Somatic SNSs and indels in CML progression Supplementary Table 3: Primers for ASXL1, IKZF3 and TP53 screening Primer name ASXL1-13F ASXL1-13R ASXL1-13aF ASXL1-13aR ASXL1-13bF ASXL1-13bR ASXL1-13cF ASXL1-13cR ASXL1-13dF ASXL1-13dR ASXL1-13eF ASXL1-13eR ASXL1-13fF ASXL1-13fR ASXL1-13gF ASXL1-13gR IKZF3-2F IKZF3-2R IKZF3-3F IKZF3-3R IKZF3-4F IKZF3-4R IKZF3-5F IKZF3-5R IKZF3-6F IKZF3-6R IKZF3-7F IKZF3-7R IKZF3-8F IKZF3-8R TP53-2-3F TP53-2-3R TP53-4F TP53-4R TP53-5F TP53-5R TP53-6F TP53-6R TP53-7F TP53-7R TP53-8F TP53-8R TP53-9F TP53-9R TP53-10F TP53-10R TP53-11F TP53-11R Sequence (5'-3') GGACCCTCGCAGACATTAAA AGCTCTGGACATGGCAGTTC GGTCAGATCACCCAGTCAGTT CACCACCATCACCACTGCT CAACTACTGCCGCCTTATCC GTCGGTGAGGATTCAGGTGT TGAAGGATCCTGTAAATGTGACC ATTGCTGTCACTGCCTCCTC TCACTCTGGACTGTGCCATC GCAGCAACTGCATCACAAGT CTCAGTGGAGGCCACTAACC CATAGCACGGACTTCCTTCTG CAGAAGGAAGTCCGTGCTATG GGGACTATGCCCAGTAGCTTT GGGTCCTCTTAAGGCAAATG GTTTCCCATGGCCATAATTT GCTGTTAAGTTTTCCGAAATGG AAGCACATTCCTCTCTCTTTGG TGTGTGAAGCTGAAAGTTAAATGG CTACAATTGCAAGTTTTCCTTGG TCGTTGTTCATTTCTTGCATTT TTCTCACGTGGCTGCATTAG GAGCTTTTCCCCTAGGCATC ACACTGGGCTCTGAGGAATG AGTTGTCACAGAGCCCCAAT ATCTGTCTGCCCCCAGTAAA GCATCCAACTTCGGACACTT TTTAACAGAGGTTAAGCTAGGAAAGG CTTGGCAGTGTTCCCTTTGT GGCGAGGTCATTGGTTTTTA CCATTCTTTTCCTGCTCCAC TCAAATCATCCATTGCTTGG CCTGGTCCTCTGACTGCTCT GACAGGAAGCCAAAGGGTGA GTTTCTTTGCTGCCGTCTTC GAGCAATCAGTGAGGAATCAGA CTGCTCAGATAGCGATGGTG TTGCACATCTCATGGGGTTA GCACTGGCCTCATCTTGG GGGATGTGATGAGAGGTGGA TCTGGCTTTGGGACCTCTTA GGAAAGAGGCAAGGAAAGGT AAGCAAGCAGGACAAGAAGC TGTCTTTGAGGCATCACTGC AACTTGAACCATCTTTTAACTCAGG GAAGGCAGGATGAGAATGGA AAAGCATTGGTCAGGGAAAA GCAAGCAAGGGTTCAAAGAC Supplementary Figure 1: Karyotype during CML progression (A) chronic phase: 45, XY, t(9;22)(q34;q11.2), rob(13;14)(q10;q10) [20]; (B) complete cytogenetic response: 45, XY, rob(13;14)(q10;q10) [20]; and (C) blast crisis: 44, XY, t(9;22)(q34;q11.2), rob(13;14)(q10;q10), -17 [5]; 45, XY, t(9;22)(q34;q11.2), +10, rob(13;14)(q10;q10), -17 [5]; 46, XY, t(9;22)(q34;q11.2), +8, +10, rob(13;14)(q10;q10), -17 [3]; 47, XY, t(9;22)(q34;q11.2), +8, +10, rob(13;14)(q10;q10), -17, +19 [2]; 45, XY, rob(13;14)(q10;q10) [5]. Supplementary Figure 2: Sequencing workflow and bioinformatics pipeline for identification of somatic mutations. After alignment of the sequencing reads from DNA samples of CML and normal cells to the human reference genome, a series of filters were applied to discard reads that were not usable for the downstream purpose of somatic mutation discovery. Sequence variants fulfilling the additional criteria were subjected to Sanger sequencing validation. References 1. 2. 3. 4. 5. 6. Rubio-Camarillo, M., et al., RUbioSeq: a suite of parallelized pipelines to automate exome variation and bisulfite-seq analyses. Bioinformatics. Li, H. and R. Durbin, Fast and accurate short read alignment with BurrowsWheeler transform. Bioinformatics, 2009. 25(14): p. 1754-60. Homer, N., B. Merriman, and S.F. Nelson, BFAST: an alignment tool for large scale genome resequencing. PLoS One, 2009. 4(11): p. e7767. DePristo, M.A., et al., A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 43(5): p. 491-8. Jones, S., et al., Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 330(6001): p. 228-31. McLaren, W., et al., Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 26(16): p. 2069-70.