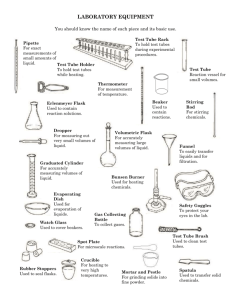
LAB #1: WATER PURIFICATION AND
advertisement

Lab Manual For BITC 140206 Spring 2006 Linnea Fletcher PhD 1 Table of Contents Introduction, Lab Safety and Lab Notebook………………………………...3 Lab #1: Water and Cleaning Glassware …………………………………..12 Lab #2: Preparation of Solutions…………………………………………..20 Lab #3 Dilutions and LAL Endotoxin Test………….……..……………..27 Lab #4: Biphasic Separation of Organic Molecules……………………….36 Lab #5: Reduction of a Carbonyl by Fermenting Yeast…………………...41 Lab #6: Synthesis of Banana Oil, an Ester………………………………...48 Lab #7 Protein Determination…………………………..………………...51 Lab #8: Preparation and Use of Media …………….………..……………58 Lab #9: Acid Phosphatase Kinetics and Inhibitors………………..………63 Appendix A: Mandatory laboratory Safety Rules…………………………73 2 Introduction to BITC 1402 Lab The Biotechnology 1402 course at Austin Community College is considered a first year course in the Biotechnology program. It is designed for both certificate and degree students who have completed or are co-enrolled in the Introduction to Biotechnology 1311, and have had at least one semester of General Chemistry and one semester of Cell and Molecular Biology for Majors, 1406 or equivalent. The objectives of the lab portion of the course are to: Develop the basic laboratory techniques of chemistry and biochemistry Supplement and enrich the lecture portion of the course, which deals predominantly with bioorganic chemistry and biotechnology lab skills. Elucidate some of the potential careers in biotechnology that are available Develop critical thinking skills in the students Encourage teamwork and accountability among the students Practice accuracy in calculations and in writing scientifically Develop multitasking skills Encourage students to take charge of their learning Learn the responsibilities associated with working in a company Students are expected to behave professionally at all times. Lab notebooks will be maintained and graded in the lab. The lecture is to give background and relevant information about the solutions, prep, procedure and related techniques. Lab is to be mainly conducted on Wednesdays except when more time is needed to prep for a lab (e.g. preparation of solutions). Equipment Responsibilities at a Company Biotechnicians are required to use, maintain, and troubleshoot a variety of different types of equipment. As part of your lab experience, you will learn these skills during the lab lectures and practice them in the laboratory. The Lab Notebook: Each student will maintain a lab notebook. The lab notebook will contain the weekly pre-lab, lab notes and protocols including all data and observations, and the post-lab analysis and questions. Note, this notebook is a learning tool and therefore contains more information than some laboratory notebooks required by industry. In industry, a technician, in the notebook, refers to a numbered Standard Operating Procedure or SOP to perform a task, or fills out a worksheet, only noting deviations from the SOP and not writing the whole protocol down again. However, we have found, that students better understand the laboratory exercise if they prepare a prelab and also dissect the laboratory exercise in their lab write up. 3 The Prelab must be prepared before beginning the lab. The lab instructor will sign off on the prelabs at the beginning of class to ensure that the students are prepared for the lab. The Pre-lab preferably should be written on the computer and consists of: A Title Purpose of the lab or scope: What is/are the purpose(s) of the lab? Include the reaction equation if applicable. For example, provides the method for assessment of Endotoxin in a solution. Table of materials and reagents: For the starred items (*), include its purpose in the lab procedure. If it is a reagent, give its name, when it was made and by whom and a brief description of its function and, if it is composed of more than one chemical, what each chemical does in the procedure. Hazard Communication: Give all the safety precautions for the lab, both those listed in the manual and those on the MSDS sheets for the chemical used in the lab. For example, if you use Hydrochloric Acid, state the following, which is part of the MSDS and also found on the bottle: Hydrochloric Acid, HCl DANGER: Corrosive. Avoid contact with skin and eyes, Avoid inhalation of fumes and mist. Do not mix with caustics or other reactives. Flowchart (optional): This chart should only include, in a step-wise manner, what the steps are in the procedure. The chart provides a quick visual of what you will be doing in the lab. Your laboratory instructor will indicate when this is required. Protocol: Write the protocol in your own words on the left-hand side of the page. Leave the right-hand side of the page for lab notes. Calculations, even though done on the calculator, must be written out in the protocol. Data tables should be copied from the manual or prepared beforehand. Your instructor may provide you with an electronic copy of the lab. BUT you should alter it to indicate that you have CAREFULLY reviewed it and changed it to fit your needs. Protocols copied from the lab manual word for word will NOT be accepted. Students who do not have a prelab will be directed to prepare one before proceeding with the lab. Lab Data: During Lab, the student is expected to take notes and record the information in the laboratory exercise they will be handing in for a grade, and not on a paper towel etc. How much exactly was weighted for each reagent? How much water exactly was used? Who did the measuring? What time did the mixture start boiling? When did the distillation finish? What mistakes were made? What was the color? AND remember to record equipment identification number and SOP number for equipment validation, if available (a big part of your lab grade will depend on writing down your observations and data.). This must be done in pen. Absolutely no pencils allowed. If you need to correct, cross it out, make the correction, initial and date it. 4 Post-Lab Results and Discussion: Calculations involving the data must be included for at least one series of measurements and proper statistical analysis must be included in this section. For many experiments, the clearest presentation of data is in a tabular or graphical form and directions may or may not ask for this type of presentation so you will need to think about the best way to present the data and do so in your lab report. Everything needs to be recorded as neatly as possible in your notebook. Discussion is the most important section of your write-up, because it answers the questions, “Did you achieve your proposed goals and objectives?” and “What is the significance of the data?” Any conclusions that you make must be supported by experimental results. If it is possible to compare your data to controls or known values obtained from reliable sources, then calculate the percentage error and why there are differences. If problems were encountered in the experiment, these should be outlined with possible remedies for future experiments. This is also a good place to list laboratory tips that have been given to you or you have discovered. References: All library references (books, journal articles, and Web sites) that were used to write up the experiment should be listed at the end. 5 Safety in the Laboratory Complete the 3 Assignments BEFORE LEAVING THE LAB Objectives Your performance will be satisfactory when you are able to Discuss safety rules for the laboratory Recognize the correct procedure for storing and handling hazardous materials Find information on the classifications of chemical hazards, what types of health hazards a chemical may pose, what levels of medical attention are required following exposure to a hazardous chemical, and what personal protective equipment is required for handling a hazardous chemical Locate the lab safety equipment Locate online Material Safety Data Sheet (MSDS) databases Locate the supplies for your lab exercises Biotechnology laboratories are equipped with supplies and equipment that may pose a hazard if used carelessly and it is important that you learn how to handle them properly. It is often the responsibility of a biotechnician to make sure that safety rules are followed, and anyone working in a laboratory must pay attention to what they are doing and use common sense to avoid hazardous situations. While the ACC science safety rules are designed to provide protection to you while working in ACC laboratories, you must become self-sufficient in protecting yourself in your future jobs in the biotechnology industry. In addition, lab technicians are frequently entrusted with ensuring compliance with safety precautions in the biotechnology workplace. For this purpose, this lab exercise will introduce you to key components to lab safety precautions and procedures that apply in a biotechnology setting. 1. Proper handling and storage of chemicals and reagents There is no single simple formula for working safely in the laboratory, since each lab facility and each experiment presents unique challenges. We will be addressing safety issues with each experiment that we do in this course and give you some specific guidelines for safety throughout the semester. A. MSDS (Material Safety Data Sheets) While each chemical that you use will have its own unique properties, there are some common practices that will aid you in treating them all with the level of respect that they are due. For example, labeling each chemical is required under the law and should be thorough enough so that even a person who does not work in the lab can identify any chemical. Also, every chemical in the laboratory should have a Material Safety Data Sheet (MSDS) on file and readily available. The MSDS is a legally required technical document, provided by chemical suppliers, that describes the specific properties of a chemical. Besides the MSDS on file in the lab, several web sites offer MSDS databases. They are all broken down to the same 8 sections: 1. Chemical identity. The manufacturer’s contact information is here, along with contacts for emergency situations. 2. Hazard ingredients/identity. Some reagents have multiple components, and many singlecomponent chemicals have alternative names. These are all listed here. Concentration limits for airborne exposure to a chemical are listed here. Although these indices of toxicity are mainly of concern for production workers in factories, they are also useful for evaluation of short-term exposures. The TLV (threshold limit value) is the maximum airborne concentration of a substance to which workers can be repeatedly exposed without adverse effects. The units used are usually parts per million (ppm) or mg/m3. 6 3. Physical chemical characteristics. This list of physical properties tells you whether the chemical is solid or liquid and how volatile it is. 4. Fire and explosion hazard data. This is of particular interest in cases where fire-fighting methods must be selected. 5. Reactivity data. This information is essential in determining the proper handling and storage of chemicals. By knowing the reactivity patterns of a chemical, you know what substances or conditions from which you must isolate the chemical. For example, acids and bases react with each other rapidly, giving off large amounts of heat, so should not be stored next to each other. Others react with water and should be stored in sealed containers with desiccants. 6. Health hazards. The best source of specific toxicology data is given here, such as symptoms of acute damage from exposure and some recommended emergency procedures. If a chemical has been tested for its carcinogenicity, or cancer-causing potential, that information is listed here. In addition, levels at which a chemical has been found to be lethal (called the LD50 for lethal dose for 50% of test animals) is listed here. Since the LD50 is dependent on which type of animal it was tested on, as well as how the animal was exposed to the chemical, this information always requires these specifics. For example, the lethal dose for chemicals is much lower if injected than it is if ingested. The most common index reported is the LD50 for a rat in mg of chemical per kg of animal, administered orally (ingestion). For volatile chemicals, the toxicity of breathing it is measured as the LC 50 (lethal concentration in air for half of the test animals), measured in ppm; in all cases, the lower the number for the LD50, the more toxic the chemical. 7. Precautions for safe handling and use. This describes how to deal with spills. 8. Control measures. Specific recommendations for personal protective equipment (PPE) are given here. B. NFPA Ratings (National Fire Protection Association) Another quick assessment of a chemical’s health hazards that is usually available on its container is a rating by the National Fire Protection Association (NFPA). A color-coded diamond shape lists numbers rating a hazard as: Blue for health hazard 0 – normal material 1 – slightly hazardous 2 – hazardous Red for flammability 0 – will not burn 1 – flash point > 200o F 2 – flash point > 100o F 3 – extreme danger 3 – flash point < 100o F 4 – deadly 4 – flash point < 73o F Yellow for reactivity 0 – stable 1 – unstable if heated 2 – violent chemical change 3 – shock and heat may detonate 4 – may detonate The uncolored station of the NFPA diamond is for specific hazards: OX ACID ALK CORR W – oxidizer compound – acidic compound – basic compound – corrosive compound – use NO WATER B) General Safety Precautions in Handling Hazardous Chemicals in the Lab There are generally four routes to exposure to hazardous chemicals that you should keep in mind while handling them: Inhalation – avoid by the use of fume hoods and masks Skin & eye contact- avoid by the use of lab coats, gloves, and goggles Ingestion- avoid eating or drinking in the lab or leaving the lab without removing gloves and washing hands Injection– dispose of broken glass and needles properly 7 Because chemicals pose so many different kinds of hazards, there are no simple rules of thumb for safe handling of them all except for some common sense measures: Treat all chemicals as if they were hazardous until you learn otherwise Label all containers with contents, including concentrations and date that they were transferred If a hazardous material is contained, label it with a warning Think through your experiment BEFORE doing it, making sure that you will not be combining incompatible chemicals Clean your bench top before and after use Wash hands often and ALWAYS before leaving the lab Take off lab coats and gloves before leaving the lab Always remove gloves before touching phones, doorknobs, light switches, etc. Ensure proper waste disposal and labelin. Here are some specific tips for handling the different types of hazardous chemicals: Flammables: Do NOT heat these reagents unnecessarily, and never in the presence of a flame or source of a spark. In general, only open containers in fume hoods. When storing more than 10 gallons of flammable liquids, a special explosion proof storage cabinet is required. Corrosives: Wear personal protective equipment (PPE) such as lab coats, goggles and gloves, and always add strong acids or bases to water when making solutions. Neutralize slowly to avoid rapid generation of heat and gases. Strong acids and bases should never be stored together. Reactive chemicals: Wear PPE such as lab coats, goggles and gloves, and know the reactive properties of the chemical. Always store oxidizing chemicals away from flammable materials. Toxic chemicals: Wear PPE such as lab coats, goggles and gloves, and know the toxic properties of the chemical. When working with a dry powder, wear a mask to avoid breathing the dust. Be aware of the waste disposal procedures for unused reagents and materials that come in contact with the chemical. Here are some of the most common hazardous chemicals that you will encounter in the biotechnology lab: Carcinogens Neurotoxins Nephrotoxins Corrosives – formaldehyde – acrylamide – acetonitrile – phenol, strong acids & bases Mutagens Teratogens Hepatotoxins – ethidium bromide – formamide – chloroform Often vendors such as Fisher Scientific have safety information in their catalog about chemicals that they sell, in which case you can easily assess chemical hazards before you order a chemical. Spectrum Chemical also has a very large collection of MSDS on their website. 8 2. Biological Safety: Containment You will be working with live organisms in many biotechnology labs, so it is important to be able to assess any biological hazards that they may pose and to treat them accordingly. In general, a live organism is considered a biological hazard if its release into the environment could have an effect on the health of the environment in general or humans in particular. This includes known pathogens to humans, plants, or animals, as well as benign organisms containing recombinant DNA that could render the recombinant host dangerous. In fact, the recombinant DNA itself should be treated as a biosafety hazard, since it is usually inserted into a vector that could transform organisms in the environment if released. Similarly, tissue cultures of human or animal cells should be treated as a biohazard: while they would not survive if released into the environment, they contain recombinant DNA. The routes of exposure to infectious agents are the same as those of hazardous chemicals: inhalation, contact with eyes and skin, ingestion, and injection. The same general precautions should be taken in handling biological hazards as the guidelines above for handling chemical hazards, especially toxic ones. Here are some general practices to maximize biological safety: Limit access to the lab at the discretion of the lab director, and adequately train all lab personnel. Use personal protective equipment (PPE) at all times, and keep all PPE inside the lab. Wash hands after handling viable materials and animals, after removing gloves and before leaving the lab. Always remove gloves before touching phones, doorknobs, light switches, etc. Avoid touching your face with your hands or gloves. Keep personal items such as coats and book bags out of the lab or in a designated work area. No mouth pipetting; use mechanical pipetting devices. Minimize splashes and aerosol production. Disinfect work surfaces to decontaminate after a spill and after each work session. Disinfect or decontaminate glassware before washing. Decontaminate all regulated waste before disposal by an approved method, usually by autoclaving. Have an insect and rodent control program in effect. Use a laminar flow biological safety cabinet when available. Seventy percent of recorded laboratory-acquired infections are due to inhalation of infectious particles, so special precautions should be taken to avoid producing aerosols when working with pathogens. While performing activities that mechanically disturb a liquid or powder, the biotechnologist should make the following adjustments. Activity Adjustment Shaking or mixing liquids mix only in closed containers Pouring liquids pour liquids slowly Pipetting liquids use only cotton plugged pipets Removing a cap from a tube point tubes away when opening Breaking cells by sonication in the open sonicate in closed containers Removing a stopper or cotton plug from a culture bottle remove slowly Centrifuging samples use tubes with screw cap lids Probing a culture with a hot loop cool loop first Disinfectants such as bleach and ethanol are used extensively to decontaminate glassware and work areas, and it is important to realize that the effectiveness of disinfectants depends on the type of living microorganisms you are encountering: 9 Resistance Level Type of Organism Examples Least resistant enveloped viruses HIV Herpes simplex Hepatitis B Slightly resistant bacteria E. coli S. aureus Medium resistance fungi Candida species Cryptococcus Highly resistant nonenveloped viruses rhinovirus Polio virus M. tuberculosis Mycobacteria Most resistant spores B. subtilis spores Clostridium species 3. Disposal of Hazardous Chemicals and Biological Materials The disposal of hazardous chemicals is subject to state and federal regulations, and is ultimately overseen by the Environmental Protection Agency. Extremely toxic chemicals are regulated at low levels, and less toxic chemicals can be disposed of through city sewer systems at higher levels. Biological hazards should be contained in autoclave bags made of a high melting point plastic that are sealed and autoclaved at high temperatures and pressures to completely kill any live organisms. First Day Lab Assignment 1: Turn in before you leave the lab 1. ACC Safety Policies (also read the information given in the Appendix) You must do the following to comply with college wide safety policy: a. Watch the ACC Biology Safety video b. Read the ACC Biology Safety Policy in your lab manual c. Fill out the Biology Safety Rules and Information sheet for this laboratory classroom d. Sign the safety contract Until you complete all of the above activities, you are not allowed to attend laboratory classes at ACC. 2. Mapping the Laboratory Mark the location of: eyewash stations, first aid kit, spill response supplies, sinks, lab benches, fume hoods, fire extinguisher, windows, exits, fire blanket, emergency evacuation rally point (outside) and route to it. You will also be responsible for gathering materials you need for each lab exercise during the semester. You will need to know the location of the following, and if you don’t know what the item is or can’t find it, use the equipment locator document located in a folder on the side of the fume hood. glassware broken glass disposal gloves freezer (-20˚C) hotplate/stirrers refrigerator (4˚C) micropipetters 37 ˚C incubators micropipetter tips microcentrifuge tubes microfuges microscopes ring stands and clamps test tube racks Eppendorf tube racks marking tape You will also occasionally need to locate chemicals and reagents for your lab exercises. 10 flammables corrosives toxins buffers 3. oxidizers reactives gas cylinders enzymes Assignment 2 to be turned in: Finding MSDS and Safety Information on the Internet Use the Internet to search for chemical company websites, university departments, or other databases containing MSDS information. Locate information for the following 3 chemicals: a. Caffeine, an addictive substance found in coffee. b. Diethyl ether, a common organic chemical. c. Tris, a buffer. For each, find the LD50 (lethal dose for 50% of the population) and whether it is a mutagen or carcinogen. 4. Assignment 3 to be turned in: Special Safety Precautions for Individual Lab Exercises Find a partner to work with, and select a laboratory exercise together from this lab manual that has a list of chemicals and materials that will be used. Using information from MSDS, find the following information: chemical name (trade name) number and identity of components NFPA rating any health hazards LD50 (mg/kg, test animal, how administered) or LC50 (ppm) carcinogen, mutagen, teratogen, neurotoxin, nephrotoxin, or hepatotoxin waste disposal method any PPE needed Enter the information in the form provided in the Appendix. The simplified categories of hazardous materials found in the appendix of this manual will help you with assignment 3. Additional information can be found in the book, Basic Laboratory Methods for Biotechnology by Lisa Seidman and Cynthia Moore, chapters 28, 29 and 30. 11 LAB #1: WATER PURIFICATION AND CLEANING GLASSWARE A: Types of Water Water is the most important solvent used in the lab. The introductory readings introduce the three types of water; Type I, Type II and Type III. Type I water is the highest class of purity and is used for most analytical procedures, tissue culture and instrumentation since it has very low levels of contaminants. It is routinely prepared by reverse osmosis (RO) or distillation and deionization. Type I water is highly reactive and is stored for only brief periods of time before use AND it must be stored in containers that do not leach minerals or organic compounds. Type II water is suitable for most routine lab work and is prepared by distillation or RO. It should also be stored in nonleachable containers BUT it is not as reactive as Type I water. Type III water, tap water, is useful for some applications such as rinsing glassware or preparing microscope slides. Note, these are “loose guidelines” and in every case, laboratory water needs to be tested and purification methods tweaked (i.e. the addition of carbon filtration or ultrafiltration etc.) to ensure the proper type of water is produced; the water should be routinely tested to ensure that the quality is maintained. Water used for making pharmaceuticals requires even stricter standards than Type I water. The United States Pharmacopoeia (USP) distinguishes between water for injections (WFI) and purified water. WFI water must be produced by distillation or reverse osmosis and must have extremely low endotoxin levels. It is also the water of choice for maintenance of mammalian and insect cell lines, as they are sensitive to the presence of endotoxin. Endotoxins are pyrogens; induce fever in humans. They are part of the cell wall in gram negative bacteria, and are lipopolysaccharides—molecules containing lipids and polysaccharides. A person with a systemic gram negative infection runs the risk of dying from septic shock when treated with an antibiotic that kills the bacterial infection; as the killed bacteria release endotoxin triggering the person’s immune system resulting in septic shock. Tap water is contaminated with endotoxin and therefore NOT appropriate for injection. The concentration of endotoxin is measured in endotoxin units or EU. In lab 3 you will perform a test for the presence of endotoxin known as the Limulus Amebocyte Lysate Test or LAL test. B. Purification of Water Most labs and biotechnology industries begin with partially purified tap water, the water we drink. The quality of municipal water varies from place to place. You may have noticed how water tastes different in different cities. Some biotechnology industries even choose their site depending on the quality of the local water. Although this local water is drinkable, it needs to be further purified for lab work. There are 5 major ways in which water is purified. They are 1. Distillation, 2. Ion exchange, 3. Carbon adsorption, 4. Filtration and 5. Ultraviolet oxidation. Distillation is often used to purify water for the lab. Water is heated in a container to boiling. The steam is collected and cooled back to water. The water is 12 collected in the appropriate glass container. Although distillation removes many contaminants, a few contaminants such as carbon dioxide, chlorine, ammonia and small organic molecules are still present. Distillation is often used to treat water although it is relatively expensive because of the heat needed to vaporize the water. Stores sell distilled water for use with contact lenses, for use in irons and other common purposes. In ion exchange water passes through filters with bead-shaped resins, which remove ions. Cationic resins remove positive ions and anionic resins remove negative ions. In Central Texas, we have hard water, water in which many ions, particularly calcium, are dissolved. We can see this hard water in the low sudsing of our soaps and shampoos as well as the tough-to-remove ring around the tub. Water softeners are cationic resins with Na+ loosely attached to them. As our hard water is passed over the water softener, the hard water Ca++ ions are exchanged for the Na+ ions. Many homes use these water softeners. However, because of the presence of Na+, they are not sufficient for lab purification. Lab water must be deionized. Deionization is accomplished using both a cationic exchange column with H+ (instead of Na+) ions as well as an anionic exchange column with OH-. The positive contaminants are removed by the first column and exchanged with the H+ and the negative contaminants exchanged for the OH-. The resulting ions combine to form more water molecules and the water is purified. It is more pure than tap or softened water but still contains organic contaminants, which are nonionic, pyrogens and microorganisms. Carbon Absorption removes organic compounds from water. The water is passed over activated charcoal (carbon) made traditionally by burning wood. Most activated carbon is made from styrene beads today, since this produces a purer carbon. The organic contaminants stick to the activated carbon and are thus removed. This is usually the preliminary step before deionization. There are four types of filtration methods used in treating water. The first of these are depth filters made of sand or matted fibers. They are often used at the beginning of the filtration system to remove large debris. Microfiltration membrane filters are filters with a characteristic pore size. Water treatment filters are usually 0.20 um which filter out bacteria, though they won’t filter smaller dissolved molecules. These molecules, including most organics, are filtered out of the water with an ultrafilter whose pore sizes are smaller than the microfilters. Reverse osmosis (RO) filters remove molecules as small as 300 D molecular weight and so remove viruses, bacteria and pyrogens. They also reject ions and polar molecules such as sugars. In RO, water is passed over a special thin membrane, which retains material based both on their size and on ionic charge. Water is usually under pressure to speed up this slow process. Reverse osmosis is often used to make Type I water. The last way to purify water is by ultraviolet (UV) oxidation. Water is passed for about 30 minutes over a UV lamp with a wavelength of 185 nm. The organic compounds are oxidized to simple compounds such as carbon dioxide. A wavelength of 245 nm will kill bacteria and is sometimes used to sterilize water. A combination of these purification procedures is often used in labs. For example Type I water may first be passed over activated carbon filter and then RO filter to a 13 storage tank. Then a series of ion exchange columns may be used before a final ultrafiltration step. Different labs have different water needs. Furthermore there is no consistent terminology used in labs to distinguish between distilled and RO and deionized water. Therefore, standard operating procedures should explicitly state the source of water required. More information concerning water testing and water purification systems can be found in chapter 24 in the book, Basic Laboratory Methods for Biotechnology. C. Operating and Maintaining Water Purification Systems Labs must monitor their water purification systems to ensure that the systems are working properly. Labs usually keep logs of the date, the monitoring system(s) used and the results. There are five quality parameters that are used to monitor the water system. 1. Resistance is a measurement of the electrical current through the water. Since ions carry the current, water without ion contaminants should have a high resistance. There is usually an attached meter to read the water resistance on water purification systems. The acceptable value for Type I water is 17.0 megohm-cm. Resistance is only good for monitoring ions in water. 2. Bacterial counts are used to monitor levels of microorganisms in water. A measured amount of water is filtered and the filter placed on a plate with nutrient agar. The numbers of colonies growing are counted to give the number of colony forming units (CFU) per volume of water. Different standards give the maximum CFU permitted. 3. As mentioned in the introduction, pyrogens are tested with a Limulus Amebocyte Lysate (LAL) test. An extract from the blood of a horseshoe crab is mixed with different dilutions of water. Pyrogens will cause the blood extract to clot. The results are given in endotoxin units per milliliter (EU/ml) 4. Organic carbon contaminants can be monitored with instruments such as the mass spectrometer, which analyze the carbon compounds. 5. The pH of ultrapure water exposed to the carbon dioxide in air is about 5.7. In a covered container, water has a pH of about 6.0. This may be monitored. The maintenance of the water purification systems include cleaning—usually daily for distillation systems, sanitation and regeneration of ion exchange systems, sanitation and checking for holes of filtration systems, and cleaning and recharging of activated carbon. D. Operation of the pH Meter The definition of pH is the negative log of the hydrogen ion (H+) concentration when concentration is expressed in moles per liter. For example, the H+ concentration of pure water is 1 x 10-7 moles/l, thus the pH = 7. By definition, any solution with a pH < 7 is acidic and any solution with a pH > 7 is basic. In this laboratory pH is measured either by using a paper pH indicator or by the pH meter. The pH meter measuring system consists of a voltmeter that measures voltage, two electrodes and the sample that is being measured. When the two electrodes are immersed in a sample, they develop an electrical potential (voltage) that is measured by the voltmeter. For more information on pH and 14 conductivity meters and their use, please read chapter 18 and the appropriate Appendix in the book, Basic Laboratory Methods for Biotechnology to review the use of the pH Meter. E. Cleaning Glassware Cleaning glassware for the lab is a little more involved than just sticking it in the dishwasher. First, there is the consideration of what items are to be cleaned. Beakers and large flasks are cleaned differently than pipettes, for instance. Secondly, the material dirtying the glassware will depend on how it’s cleaned. A round-bottom flask that had been used to cook an organic reagent will be much dirtier than a graduated cylinder used to measure out a quantity of dilute sodium hydroxide. Thirdly, the application for which the glassware will be used next must also be considered. Will the glassware be used for making solutions or growing cells? The five steps to washing glassware (or plasticware) are 1. Prerinse: Soaking or prerinsing all glassware after use. This will help prevent the contaminants from drying onto the glassware. 2. Contaminant Removal: For example, washing, using detergents and/or solvents along with scrubbing and jets of water. Typically, a lab glass detergent such as Alcon is used. For organic chemistry use, the vacuum grease and most organic reagents are removed with a little alcohol and/or acetone. As with all washing, hot water is preferable to cold water. Often lab brushes are used to help wash glassware. 3. Rinse: Removing the detergent and cleaning solvents. Many SOP’s specify that glassware should be rinsed 3 to 5 times in tap water. 4. Final Rinse: Use purified water to rinse away the final residue. This is usually done 3 times. 5. Drying: This is either done in the air on a rack or by heat. (RNA labs traditionally bake their glassware to destroy RNA-degrading enzymes (RNAses) or use commercially available solution to get rid of RNAses.) Glassware should be dried upside down so no contaminants from the air fall in. Clean dry glassware is often stored covered in closed cupboard to avoid contamination. 15 #1: Lab Protocol A. Materials Water purification system(s) Masking tape PH meter and standards Marking pens Organic lab kit Conductivity meter Lab detergent Nutrient agar plates (2 per student) RO water, distilled water, tap water grid filter paper and forceps Vacuum flask, buchner funnel, vacuum pump and hose Filter papers (0.2 or 0.45 um) B. Protocol Part I: 1. Find the two water purification system(s) that this lab uses, both the RO and distillation system; also locate a tap water source. 2. Collect 100 ml of the purified water from the systems in the lab, using a 100 ml graduated cylinder, put sample into a beaker. As directed by laboratory SOP for the pH meter, calibrate pH meter, and determine the pH value of your solutions. If available, determine the conductivity of each water sample. Record the information as indicated below. pH and conductivity of RO water : ___________________________________ Date recorded:___________________ Names of testers:__________________________________________________ pH and conductivity of distilled water: ________________________________ Date recorded: ___________________ Names of testers: __________________________________________________ pH and conductivity of tap water: _____________________________________ Date recorded: ___________________ Names of testers: __________________________________________________ 3. Filter each 100ml sample using the grid paper, buchner funnel, and vacuum pump setup, your instructor will help you. Using forceps, place the paper face up on a nutrient agar plate. Label the plate on the bottom with your name and date, place in a 25 to 30degree incubator until next lab period. If any, count and describe the colonies, both fungal and bacterial, that grow within that time frame; report CFU/ml of water. 4. When finished, clean up. Part II: 1. Take an organic chemistry kit. Using a piece of masking tape and a marking pen, record the names of partners on the kit. (DO NOT write directly on the kit.) This will be your assigned kit for the rest of the semester. 16 2. Identify the pieces of glassware using the guide provided below or found in the kit. 3. Wash each piece of glassware using the procedure described in the introduction. What kind of detergent did you use to clean the glassware? __________ Dry the outside and replace in your kit. Return your kits where you found them. 17 EQUIPMENT COMMONLY USED IN THE ORGANIC CHEMISTRY LABORATORY 18 C. Results and Analysis 1. Does the pH of the purified water available in the lab fall between normal values? If not, speculate why. 2. Why did you incubate the nutrient agar plates at 25 to 30oC? Did the filtered water samples contain bacterial or fungal colonies? If yes, describe them . Count the number of colonies and report the number per ml of water.. 3. Based on the purification systems, found in the laboratory, what are the possible types of water available in the lab? What additional tests need to be conducted to verify your water type selections? 4. Did you use any organic solvents such as acetone to clean your glassware? Explain why you did or did not. 19 Lab #2: PREPARING SOLUTIONS Introduction A major job of any biotechnician is the preparation of solutions (i.e. solute dissolved in a solvent = a solution),AND accuracy is of the utmost importance. An incorrectly prepared solution can destroy months or years of hard work. Therefore, several safeguards are in place to ensure that mistakes are minimized. First, all calculations are written down in the lab notebook, even though a calculator is used to do the arithmetic. Second, important calculations are double-checked by another person (and sometimes triple-checked). Third, the exact mass or volume of each reagent used in making the solution is recorded in the lab notebook. Fourth, this information and more, is recorded on a data sheet and on a label on the bottle itself. You will follow this protocol for every solution you make in this course. Making Solutions—Calculations Solution calculations are one of the primary reasons biotechnicians need good math skills. Review chapters 9, 16, 21, and 22 in the Basic Laboratory Methods for Biotechnology by Lisa Seidman and Cynthia J. Moore if you do not remember your chemistry math; briefly, always determine what is needed and what is given, and, when doing the problem, always write down the units and cancel them to make sure you end up with an answer that is in the correct units. Weight per volume is the simplest way of expressing a concentration. This is often used for small amounts of chemicals and specialized biological reagents. For example, enzyme concentrations are often given as weight per volume--2.0 mg/ml bovine serum albumin. Molarity is by far the most commonly used unit of concentration in the lab. Molarity is the number of moles of a solute that are dissolved per liter of solution. It is given as M. In order to calculate molarity, you need to know moles. A mole is a standard number of molecules, but since different molecules have different weight, we need to know how much a molecule weighs—it’s molecular weight (MW) or formula weight (FW). The FW may be obtained from the bottle of reagent or by adding up the atomic weights of each of the atoms in that molecule. Percents may be (1) weight per volume percent, which is the grams of solute per 100 ml of solution (w/v), or (2) volume percent, the volume of solute per 100 ml solution (v/v). Each of these concentrations has calculations associated with it. In doing word problems and in determining the correct equation, there are certain key words to look for. These key words will indicate which equation to use. The following table gives the key words, the equation and examples for each of the concentrations above. 20 Type of Solution key words to look for equation to use Weight per volume weight (mg, ug) and volume (ml, ul) __?__ = known wt given wt known vol Molarity weight/volume (M or moles/liter) ? g = F.W. x M x vol ? g = grams x mole x L mole L Percent % symbol __?__ = known wt given vol 100 ml example How many ug DNA [?] to make 50 ul [given wt] of 100 ug/ul [known wt/known vol] solution? __?__ = 100 ug_ 50 ul 1 ul ? = 100 ug x 50 ul 1 ul ? =5000 ug= 5.0 mg DNA How many g NaOH (FW = 40.0 g/mole) are needed to make 2 L of a 0.8 M solution? ? g = 40.0 g x 0.8 mole x 2 L mole L ? g = 64.0 g NaOH needed How many g NaCl needed to make 200 ml of a 15% salt solution? ____?___ = 15 g 200 ml 100 ml ? = __15 g__ x 200 ml 100 ml ? = 30 g NaCl needed * the units in the examples are not cancelled, please do this now. REMEMBER! Check your units. You may need to convert in order to have all your units cancel. Conversions: (These may be used as written or upside down so that your units all cancel out) __1 g___ or __1 g___ or __1 mg__ 1000 mg 1,000,000 ug 1000 ug 6 (or 10 ug) The same relationships hold with volumes and moles. Strategies for Making Solutions in Lab The buffer you are preparing consists of the following ingredients: 50 mM Tris, pH 7.5 1 mM MgSO4 2 % (w/v) NaCl We will be making 100 ml of this solution by two strategies. The first strategy is to prepare the buffer without stock solutions. The Tris is prepared and pH adjusted but not brought to volume (BTV) with purified water; the rest of the solutes are then added. Finally the buffer is BTV and the pH checked. 21 The second strategy involves making concentrated stock solutions and combining them to form the final concentration of each and the correct volume. Each of these two strategies are frequently used in the lab. Using a Balance Review chapter 15 in the textbook, Basic Laboratory Methods for Biotechnology, to review the proper way to use a balance. General guidelines to follow when using balances are as follows: The amount being weighed determines what type of balance is used in this laboratory (i.e. pan or analytical). Use clean spatulas to weigh out material Never put excess chemicals back into their original containers, discard Leave a CLEAN balance, chemicals left on the balance will corrode it Turn off the balance when not in use You should also refer to the SOP for whatever balance is being used. 22 Lab #2: Protocol A. Materials Trizma base MgSO4 NaCl pH meter small beakers graduated cylinders stir bars/stirrers ice bath 60oC bath conc HCl B. Hazard Communication Hydrochloric acid (HCl) can burn skin and clothing. Be careful when handling. If any acid contacts skin immediately flush with lots of water. If any spills occur, wash away with lots of water and wipe dry with paper towels. (Some labs have sodium bicarbonate available to neutralize acid spills.) Inform the lab instructor of all spills. C. Protocol Part I: Preparing solutions without Stock Solutions. 1. Calculate the g of Tris needed to make 100 ml of the solution previously described in the introduction, showing your work. Check calculation with lab partner. Weigh out this amount of Tris. If you have not used a balance before, check with the instructor to make sure you learn how to use it correctly. Record the exact number of g weighed out in your lab notebook. Measure about 70 ml water into a beaker and add a stir bar. Pour in the Tris and place the beaker on a stirrer. Stir to dissolve the Tris. 2. Calibrate the pH meter as you did last week. Rinse the probe and put it in the beaker of Tris. Make sure not to insert the probe too deeply. The stir bar can break it. It should be just below the surface. Turn on the stirrer and check the pH. Add a few drops of hydrochloric acid (HCl) --CAREFUL OF THE ACID-- and check again. You can keep the probe in the beaker and the stirrer going while you are adding the acid. Don’t add the acid too fast; allow time for stabilization of the pH between drops. Repeat until the pH of the Tris is 7.5. 3. Calculate the g NaCl needed. Double-check this calculation with your partner. Weigh out this amount of NaCl (record the exact amount weighed out) and add to the Tris buffer. 4. Calculate the g MgSO4 needed. The magnesium sulfate is usually in a hydrated form with 7 waters associated with it. Double-check this calculation with your partner. Weigh out this amount of MgSO4*7H2O, record the exact amount weighed and add to the Tris buffer. 5. Bring to 100 ml with water. Double check the pH to make sure it’s still at or near 7.5. Pour into a bottle. Label using the label form provided and fill out the media prep form on the next page. 6. Pour about one third of your buffer into a small labeled flask and place it in ice. Pour another third into a second labeled flask and place it at 60oC for Part III. 23 Part II: Preparing Buffer with Stock Solutions 1. The instructor will assign each lab team a different stock solution to prepare, a, b, or c. A stock solution is a concentrated solution, often 10X or ten times concentrated, that is prepared in advance. When needed, a small amount of the stock solution is diluted to the desired concentration and used. Then each team will use the stock solutions to make their own buffer. a) Stock solution of Tris buffer. Calculate the amount of Tris needed to make 100 ml of 1 M stock and double check with partner. Weigh out and dissolve in about 50 ml water. Adjust pH to 7.5 with concentrated HCl and BTV with water. Pour into a bottle, label as directed and fill in media prep form. b) Stock solution of MgSO4. Calculate the magnesium sulfate needed to make 100 ml of 10 mM solution and double check with partner. Weigh out the magnesium sulfate and dissolve in about 80 ml water and BTV with water. Pour into a bottle, label as directed and fill in media prep form. c) Stock solution of NaCl. Calculate the NaCl needed to make 100 ml of 10% solution and double check with partner. Weigh out the magnesium sulfate and dissolve in about 80 ml water and BTV with water. Pour into a bottle, label as directed and fill in media prep form. 2. To make the final solution it is necessary to combine the right amounts of each stock. To do so, use the C1V1 = C2V2 equation for each solute. We will be using the buffer recipe and making 100 ml of solution. Measure the correct amount of each stock into a graduated cylinder and BTV with water. Pour into a bottle and label. Fill out a media prep form. Part III: Temperature and The pH of Tris Buffers 1. Remove the flask of buffer from the ice bath. Record the temperature and pH. Assume that the thermometer has been validated. Do the same to the flask of buffer in the 60oC bath and the remaining buffer on your bench. Make a table of your data. 24 25 C. Results and Analysis 1. Define a buffer. (Look in your text or on the web) At what pH does Tris start to do its buffering? Is it a buffer at pH 7.5? 2. Which of the two strategies would you more likely use if you had to make this buffer every Monday? Explain your answer. 3. Attach your Media Prep Forms to your lab report and hand in next week. Make sure you include your sources of error. 4. Does the pH change or stay the same at different temperatures? Would preparation temperature of a buffer be worthwhile to include on an SOP? Why or why not? 26 Lab #3: Dilutions A. Introduction In Lab #2a we saw how stock solutions may be diluted to make a buffer. This kind of 1:10 dilution is a common way of diluting a stock solution to a working solution. There are two other types of dilutions: serial and parallel. Parallel dilutions are similar to the one you did last week. A stock solution may be diluted to different concentrations in different tubes. Say, for example, that you wanted to make 1.0 M Tris buffer pH 7.5; 0.5 M Tris buffer, pH 7.5; 0.1 M Tris buffer pH 7.5; and 0.01 M Tris buffer pH 7.5. You could make up the four different solutions but this would take some time. A faster method would be to make a stock solution of 2.0 M Tris buffer, pH 7.5. Then remove the correct amount from the stock and BTV to make the final solutions. This is parallel dilution as illustrated below. The stock solution is the only source of the buffer. Fig. 3-1: Parallel Dilutions 27 The other kind of dilution is the serial dilution. In this type of dilution, an aliquot is taken from the stock bottle and place in the first tube which is then BTV to achieve the correct concentration. Then an aliquot is removed from the first tube and placed in the second tube and BTV. The second tube serves as the stock for the third tube and so on. Thus a serial dilution uses the original stock solution only once. All other samples are taken from the previous tube. This is illustrated below. Fig. 3-2: Serial Dilutions B. Preparing Solutions by dilution When diluting a stock solution, either by parallel dilutions or serial dilutions the equation to use is: C1 V1 = C2 V2 This means that the Concentration of the stock times Volume of the stock used = Concentration of the dilute solution times Volume of the dilute solution. You will usually know the concentration of the stock solution, as well as the concentration and the volume of the dilute solution. Therefore, what you usually need to find is the volume of the stock solution you need to make the more dilute solution. Another way of writing this equation is: ? volstock = Concdilute x Voldilute Concstock (Choose whichever equation you like to do the calculations; they are both the same.) For example, say you need to make 10 ml of 0.5 M solution from a stock of 2.0 M. How many ml of the stock would you use? Plug in the values into the equation ? ml stock = 10 ml x 0.5 M ? ml stock = 2.5 ml 2.0 M 28 Note that the Molarities cancel out and we are left with ml. Thus we can take 2.5 ml of the stock 2.0M solution and place it in a tube. BTV with water and we will have 10.0 ml 0.5 M solution. Instead of BTV, biotechnicians often will add the appropriate amount of water to the tube beforehand and then add the stock. This saves time and glassware. How do we calculate the appropriate amount of water? We know that we eventually want 10 ml of dilute solution in the tube. Our calculations indicate that we will be adding 2.5 ml of the stock. Therefore we will need 10 ml minus 2.5 ml, or 7.5 ml water to BTV. C. Practice Problems [Do these as part of your prelab] 1. Make 3 tubes of 6 ml each of 0.4 M, 0.2 M and 0.1 M from a stock of 0.8 M. Would you use parallel or serial dilution for this? 2. Make 4 tubes of 5 ml each of 1.0 M, 400 mM and 100 mM from a stock of 2.0 M. Would you use parallel or serial dilution for this? 3. Make 1 tube of 10 ml of 500 mM and 1 tube of 10 ml of 500 M from a stock of 1.0 M. 4. Make 4 tubes of 10 ml each. Make each 1/10 as concentrated as the previous one. The stock is 2.5 mM and the first tube is 10% of the stock. D. The Micropipetter The micropipetter or micropipet is one of the biotechnologist’s most frequently used tools. There are different brands of micropipetters such as Brinkmann, Labsystems or Rainin. Each of the brands has their devotees but all of them work in the same way. All micropipetters are essentially long tubes with a handle. A disposable tip is placed on the bottom of the pipetter or pipet or micropipettor (you will see it written these three ways). This tip is the only piece that is inserted into the liquid. In or near the handle is a screw that adjusts the volume of the pipetter. On top there is a plunger button for filling and for dispensing the liquid. There is often a second button on top for ejecting the tip. Your instructor will review with you how to correctly pipet solutions, including difficult solutions such as organics or viscous solutions, and how to estimate small volumes using a micropipetter. To ensure your success at measuring small volumes, always watch the micropipetter take up the nominal volume and deliver it. Micropipetters come in a variety of sizes. The sizes we will be using in this program are varied, for example, 1-20 L, 20-200 L, and 100-1000 L micropipettes. When choosing which pipetter to use, the rule of thumb is to choose the smallest size that can deliver the desired volume. 29 A biotechnician is usually issued a personal set of micropipetters and is responsible for cleaning and verifying them on a regular basis. Calibration of the pipetters is authorized by the companies and is not done in the field. In the first section of this lab you will verify a pipetter. Read Chapter 16 in the textbook, Basic Laboratory Methods for Biotechnology by Lisa A Seidman and Cynthia J. Moore for more information on pipetting techniques. 30 Lab #3: Dilutions A. Materials Part I: micropipetters Balance, weight boat Part II: Per Group CSE (certified standard endotoxin) containing 0.5ug standard endotoxin LRW (LAL reagent water) Limulus Amebocyte LAL with a label sensitivity of 0.03EU/ml Depyrogenated soda lime test tubes micropipetters Endotoxin free pipet tips parafilm test tube rack 37°C water bath B. Protocol Part I: Verifying a Micropipette 1. Set the pipetter to ½ its maximum volume. Place a weigh boat on a balance and tare to zero. Assume that the balance has been calibrated and validated. 2. Put a tip on the pipetter, draw up the nominal volume (desired volume) of water and weigh it. Record the weight. Record the identification number for the balance. Tare the balance and repeat 9 more times, for a total of 10 weights. 3. Repeat this procedure for two more volumes, one low and one high for the pipetter. For example, a low volume for a 200 – 1000 ul pipetter is 200 ul and a high volume is 1000 ul. Table 3-1: Pipetter Verification Set Volume Weights Mean Weights 4. Find the mean of the 10 weights and record. 5. Convert the mean water weight to the mean volume measured assuming the density of water is 0.9982 g/ml at 20 C. 31 6. Following the directions in chapter 16, determine the % accuracy or the “inaccuracy”, and the precision of pipetter. Report the SD and the CV for all three volumes in the following table Volumes % Mean Error Mean Error Precision (CV) Precision (SD) Part II. Test for Endotoxin: A Serial Dilution Problem Verification of LAL Sensitivity – Gel Clot Method Note, this SOP was written by a technician who teaches in the New Hampshire Technical College’s Biotech Program. Its format has not been changed for this course. At the end of the SOP you will find listed the technician’s name who originally wrote it, and the years with the initials of the individuals who made changes to it. In a company, any proposed changes in an SOP are reviewed before they are accepted. If the SOP is regulated by FDA guidelines, proposed changes must be submitted to the FDA; most likely a time consuming, and therefore monetary consuming, step. 1. Purpose Prior to conducting a LAL test, the QC technician must first verify the sensitivity of the LAL reagent with a certified standard endotoxin. This test involves determining the endpoint (last positive dilution) using a series of endotoxin concentrations (serial dilutions) that bracket the labeled sensitivity. The labeled sensitivity must be within a two-fold dilution of the endpoint for the LAL to pass verification. This is an extremely important test in the pharmaceutical industry and this SOP is a good example of what an SOP looks like in this industry. This exercise will also test the students’ aseptic technique which is critical to prevent the introduction of extra endotoxins into the test. 2. Scope To describe the appropriate operating instructions to perform the LAL sensitivity verification test. 3. Responsibility It is the responsibility of the course instructor/lab assistant to ensure that this SOP is performed as described and to update the procedure when necessary. It is the responsibility of the students/technicians to follow the SOP as described and to inform the instructor about any deviations or problems that may occur while performing the procedure. 4. Tools are recorded in Materials for this lab. 32 5. Protocol Instructor performs steps 5.1 to 5.5. Students start at step 5.6 5.1.0 Reconstitute the certified standard endotoxin (CSE) with 5ml LAL water (LRW) to give 100ng/ml. 5.1.1. Cover the stopper with parafilm and vortex for approximately 1 minute. 5.1.2. Let the vial stand for 30-60 minutes, vortexing for about 1 minute at approximately 10 minute intervals. 5.2. Record the potency of the CSE from the certificate of analysis for the LAL/endotoxin lot combination. 5.3. Determine the number of EU/ml for the CSE by multiplying the concentration (100ng/ml) and potency. 5.4. Showing students, serially dilute the CSE with LRW to obtain a final concentration of 0.125EU/ml. See instructor on how to perform this dilution 5.5. Reconstitute the Limulus Amoebocyte Lysate (LAL) by adding 5.0ml LRW. Swirl occasionally until completely dissolved (about 3 minutes) 5.6. Set up 12 reaction tubes in a rack (2 rows with 6 tubes in each row). Label the tubes in each row as 2, , ½ , ¼, negative control, and water sample. NOTE: KEEP ALL TUBES COVERED WITH PARAFILM WHEN NOT IN USE. 5.7. Add 100ul LRW to all tubes in each row 5.8. Add 100ul of the 0.125 EU/ml standard to all the 2 tubes so that they will now contain 0.0625EU/ml. Change tips after each addition. 5.9. Vortex mix the 2 tube for 4 seconds. While mixing, put on a new tip on the pipette. 5.10. Tip the tube so that the liquid reaches the lip of the tube. Remove 100ul of the liquid and add it to the next tube in the series 5.11. Repeat steps 5.9 and 5.10 with the tube. 5.12. Repeat steps 5.9 and 5.10 with the ½ tube 5.13. Tip the ¼ tube, remove 100ul and discard it. Do not add anything to the negative control tube. 5.15. Add 100ul of the water sample to the sample tube. NOTE: ALL THE TUBES IN REPLICATE ROW 1 SHOULD NOW HAVE 100ul OF LIQUID. 5.15. Repeat steps 5.9 through 5.13 for the remaining replicate row. 5.16. Starting with the negative controls and proceeding from the lowest to the highest standard concentration, add 100ul LAL to each tube. Add 100ul LAL to the water sample tube as well. NOTE: LAL MUST BE ADDED TO ALL TUBES WITHIN 2 MINUTES OF ADDING IT TO THE FIRST TUBE AND TIPS NEED TO BE CHANGED AFTER EACH ADDITION 5.17. Shake the test tube rack vigorously for 30 seconds to mix the LAL and sample. 5.18. Cover the rack with parafilm and place it in the 37°C water bath CAREFULLY (do not disturb other racks). Check the temperature and record the time as well as starting the timer. NOTE: DO NOT DISTURB THE TUBES DURING THE INCUBATION. ONCE A CLOT IS BROKEN IT WILL NOT REFORM. 5.19. Incubate for approximately 60 minutes. 5.20. Read the test by removing the tubes one at a time from the incubator and inverting them smoothly. Score each tube as positive or negative according to whether a firm clot 33 was formed. If the gel holds, but collapses after the tube is fully inverted, score that tube as negative. 5.21. Record whether the labeled sensitivity of the LAL is within plus or minus a twofold dilution. The endpoints of the replicates should all be within a twofold dilution of each other. V1. Deb Audino NHCTC 2001 Revised 2003 dca Revised 2005 laf 34 Results and Analysis 1. Compare your micropipetting results with those results in figure 16.15, chapter 16. Is your micropipetter within the specifications indicated? 2. An experiment calls for a series of tubes containing 2 ml of 0.1 mM glucose, 0.01 mM glucose, 0.001 mM glucose and 0.0001 mM glucose. You have a stock of 1.0 mM glucose. How would you prepare these dilutions? 3. You receive an endotoxin standard solution of 7,654 EU/ml and it needs to be diluted to 0.125 EU/ml total volume 100 ul. Show how you would do this dilution given endotoxin-free 3 ml tubes and pipette tips. 35 Lab #4: Biphasic Separation Introduction to Biphasic Separation: Organic molecules that are larger than 4 carbons long are not soluble in water. The nonpolar carbons, even when attached to polar atoms such as oxygen or nitrogen, are unable to intercalate into the water molecules. All this changes, however, if an organic molecule becomes an ion. If the molecule can gain or lose a proton—act as a Bronsted-Lowry acid or base—then it becomes a charged ion and will become soluble in water. For example, benzoic acid, C6H5COOH, is insoluble in water. Although it has two polar oxygens, it has seven carbons, too many for solubility. But as its name implies, benzoic acid can donate a proton to a base: C6H5COOH + NaOH (aq) C6H5COO- Na+ + H2O Insoluble Soluble The resulting ion, C6H5COO- with its counterbalancing sodium ion is now soluble in water as are all ions. This ion, a conjugate base of the carboxylic acid, is called a salt. Its name is benzoate, the “–ate” suffix referring to the “COO-“. We can take advantage of the relative solubilities of these large organic molecules and their salts if we are faced with a situation in which we have to separate a mixture of molecules. In part I of this lab, we are presented with a mixture of benzoic acid, that can make salts, and a large organic molecule, naphthalene, that cannot make salts. Our job is to separate out the individual components of this mixture. To do this, the mixture is first dissolved in an organic, nonpolar solvent and then poured into a separatory funnel. Sodium hydroxide is added, the funnel is shaken, allowing the two immiscible layers to mix enough to convert the benzoic acid to its salt, benzoate. The salt dissolves in the aqueous layer. The organic layer retains the naphthalene. The funnel is kept stationary allowing the nonpolar and polar solutions to visually separate. Physically separate the two layers. Now that we have separated the two components of the mixture; we need to recover the naphthalene and benzoic acid from their environments. The best way to do that is to make them insoluble again. Acid is added to the benzoate extract causing the benzoic acid to reform and precipitate out so it can be filtered. The naphthalene may be isolated from the organic solvent by evaporation. 36 Lab #4: Protocol A. Materials For Part I: Mixture comprised of 1:1 benzoic acid and naphthalene diethyl ether 2.5 M NaOH 3 M HCl ring stands and clamps red and blue litmus paper Buchner filter, filter flask and vacuum tubing Filter paper Balance Organic kits (should have a separatory funnel) 250 ml beakers Ice Part II: Set-Up for Next Week Sucrose Methylacetoacetate Baker’s yeast Warm tap water Sterile 125-ml Erlenmeyer flasks parafilm Dibasic sodium phosphate 30oC incubator B. Hazard Communication The benzoic acid, and naphthalene are organic reagents. Avoid contact with the skin, the eyes or respiratory system. If any gets on skin, wash with soap and water. Flush eyes with water. Inform the lab instructor of any accidents. Wear goggles when working with these reagents. The 2.5M NaOH is a strong base. Avoid contact with the skin washing with large amounts of water if there are spills. C. Protocol Part I: Biphasic Separation 1. Weigh out approximately 2 grams of the 1:1 benzoic acid: naphthalene mixture. Record the exact weight in your notebook. Dissolve the mixture by swirling it with 30 ml of diethyl ether in Erlenmeyer flask. If any solids remain, add more diethyl either to affect complete dissolution. Record how much solvent you used. 2. Checking to make sure the stopcock to the sep funnel is closed; pour the mixture into the sep funnel. Extract the solution by adding 15 ml portion of 2.5 M (10%) aqueous sodium hydroxide. Stopper and shake, shake, shake then vent (remove glass stopper), aiming the end away from your face and that of your partner. Repeat two more times. 3. Clamp the sep funnel to a ring stand and let sit a moment so the two layers have time to separate. Remove the stopper and place it on a paper towel. A stopper should NEVER 37 be placed directly onto a bench. Bench tops are dirty and a common cause of contamination. 4. To decide which layer is organic and which is aqueous, take the water bottle and squirt a little water into the sep funnel. If the aqueous layer is on the bottom, the water will pass through the top layer and mix with the bottom layer. Remove the aqueous layer into a clean beaker labeled “Hydroxide Extract”. 5. Transfer the organic solution into a clean Erlenmeyer flask containing two spatula-tips full of anhydrous sodium sulfate and label “Neutral Compound”. Let this solution stand for about 15 min, occasionally swirling it to hasten the drying process. If the solution remains cloudy, add additional portions of sodium sulfate to complete the drying process. 6. Cool the “Hydroxide Extract” in an ice-water bath. Carefully acidify this solution with 3 M hydrochloric acid, so that the solution is distinctly acidic to pH paper. While the acidic precipitate is cooling, weigh a piece of filter paper and record the weight. 7. Assemble a filter apparatus as follows: Take a thick-walled vacuum hose and attach to a vacuum pump. Attach the other end to the side-arm of the filter flask. Place the Buchner funnel on top. Place the preweighed filter in the funnel (It may help to sketch this in your prelab.) Turn on the vacuum. Squirt a little water onto the filter to wet it, then pour in the neutralized acid extract. Wash the solid on the filter paper with a small portion of cold water. This is your benzoic acid sample. 8. Turn off the vacuum and remove the filter to a previously weighed (tared) watchglass, cover it with a piece of filter or weighing paper, and allow the product to air-dry until the next laboratory period. 9. Separate the “Neutral Compound” from the drying agent by decantation or gravity filtration through a cotton plug into a tared 100 ml round bottom flask. Remove the solvent by simple distillation. Allow the residue to cool to room temperature to solidify. This is your naphthalene extract. NEXT WEEK: Weigh both the acid extract and neutral extract precipitates. Subtract the weights of the tared containers and calculate the percent recovery for each as: __weight of crystals recovered___ x 100 = percent recovery weight of original sample Note: To get the denominator, the weight of the original sample of acidic extract or neutral extract, go back to step 1 and calculate how many g benzoic acid and how many g naphthalene are in the original sample; remember it was a 1:1 mixture. 38 Part II: Set Up for Next Week The yeast is allowed to ferment for a week, during which time it reduces the keto ester to a hydroxy ester. Yeast is a facultative anaerobic organism and therefore, to promote fermentation, the culture needs to be grown anaerobically (without oxygen); so no shaking of the culture! 1. In a sterile 125-ml Erlenmeyer flask, dissolve 8.0 g sucrose and 0.05 g dibasic sodium phosphate in 15 ml warm (35oC) tap water. Add 1.6 g bakers yeast. 2. Swirl until all ingredients dissolved.. Add 0.5 ml methyl acetoacetate and swirl again to mix. 3. Cover with parafilm. Take a square of parafilm and hold one corner on the edge of the flask. Stretch and pull the parafilm over the top of the flask, twist and repeat. Place in the warm (30oC) incubator until next week. 39 Results and Analysis Part I: 1. Which compound is in the basic extract and why? 2. What do you think might happen to your percent recovery if you did not cool the precipitates before filtering? 3. A student calculated his percent recovery at 110%. Give at least one possibility of what went wrong. Part II: 1. Could you set up this experiment with glucose instead of sucrose? Why or why not? 2. What would be the yield of product if the yeast were kept in the refrigerator for the week? 40 Lab #5: Reduction of A Carbonyl A. Introduction Glycolysis is the process in cells by which glucose, a six-carbon sugar, is broken down to two molecules of pyruvate, a three-carbon carboxylic acid. During glycolysis, a net of 2 ATP, energy “packets” for the cell, are produced. This production of ATP is the driving purpose of glycolysis, a process performed by virtually all cells including bacteria, plant cells, animal cells, protests and fungi. A by-product of glycolysis is the formation of NADH plus 2 H+ from NAD+. At one point along the pathway, two hydrogens are removed from an intermediate molecule. NAD+ acts as the hydrogen acceptor and is reduced to NADH. If the NADH does not donate its hydrogens to another molecule, then NAD+ cannot be regenerated. Glycolysis would be halted by the lack of NAD+ and the cell would die. Not a good situation for the cell. Cells have several mechanisms to regenerate NAD+. If a cell is aerobic and breathes oxygen then the NAD+ is regenerated as part of the process known as electron transport. Cells grown without oxygen—anaerobically—will ferment to regenerate the NAD+. Yeast cells will produce ethanol and carbon dioxide as a by-product when they undergo fermentation. (The alcohol and bubbles of alcoholic beverages.) Thus, when we drink beer or any other alcoholic drink we are really drinking “yeast waste”. The pathway for yeast fermentation is: Yeast take the pyruvate from glycolysis and remove a carbon dioxide. This produces a two-carbon aldehyde, ethanal (acetaldehyde). In the second step, ethanal is reduced to ethanol using the NADH and regenerating the NAD+ (Why is it important to regenerate the NAD+ in fermentation?). We can take advantage of the fact that yeast will not only reduce ethanal; it will also reduce many other carbonyls. If we feed fermenting yeast methyl acetoacetate, a keto ester, the double-bonded oxygen—the carbonyl—is reduced to an hydroxy. 41 Last week we set up the fermenting yeast, using sucrose as the energy source. Sucrose is a disaccharide that is made of one molecule of glucose and one molecule of fructose. Glucose is the first substrate in glycolysis and fructose can be converted into glucose and also enter glycolysis. This week we will isolate the hydroxy ester product, methyl-3-hydroxybutyrate. Yeast cells are isolated by centrifugation. The pellet is resuspended in NaCl to lyse the cells and the hydroxy ester is extracted with dichloromethane. After simple distillation to remove the dichloromethane, the solute is detected by ferric chloride test. Introduction to Simple Distillation One of the oldest biotechnologies is the making of alcohol. Yeast and a food source are mixed together in an airtight container. In the absence of air, the yeast undergoes fermentation in order to continue glycolysis and the production of ATP. The by-product of this fermentation, ethanol, accumulates in the medium. The question then becomes how to separate the ethanol from the yeast and other contaminants. The answer is distillation. Different liquids have different temperatures at which they boil (change from a liquid to a gas). In general, small molecules have low boiling points, and larger molecules or molecules that can form hydrogen bonds with each other boil at much higher temperatures. Thus propane with only 3 carbons has a boiling point of –42oC and so is a gas at room temperature (propane gas is sometimes used to heat homes and in stoves and water heaters). On the other hand, the boiling point of octane with eight carbons is 126oC. Therefore octane, a major component of gasoline, is a liquid at room temperature. The boiling point of ethanol is 78.3oC. Although it is small with a formula weight of only 46 g/mole, it has an –OH group that allows it to form hydrogen bonds. This keeps the ethanol trapped in the liquid phase and raises the boiling point. If the fermenting liquid were heated, the ethanol would vaporize at 78.3oC, well below the boiling point of water (though an azeotrope, a combination of 95% ethanol and 5% water, is what actually boils off in this unique case). If the ethanol vapors are collected, then condensed back to the liquid, then the alcohol is separated from the other contaminants. This is the principle behind distillation. All mixtures of liquids with different boiling points may be separated by distillation. There are two types of distillation, simple in which there is only one or two liquids and fractional distillation for mixtures of three or more liquids. (The refining of crude oil is actually a fractional distillation of the oil into its different components.) In simple distillation there is a still pot that is heated. On top there is a thermometer to measure the boiling point and a condenser. The West condenser is a tube-within-a-tube. The outer tube of glass is called a water jacket. Cold water is passed through the outer jacket condensing the vapor in the inner tube. The liquid is collected at the end. Occasionally, the distillate vapor is cooled by air passing over a long coil of copper tubing. 42 43 Fractional distillation requires another step since there are more liquids in the mixture. A Hempel fractionating column is added above the still pot. This is a long glass tube that is often filled with glass beads or other nonreactive material. As the vapors work their way past the glass beads, the compounds with the higher boiling points recondense on the beads and return to the still pot, leaving the compound with the lowest boiling point to continue up to the West condenser. With both types of distillation the temperature of the vapor is monitored. As heat is applied to the still pot, the contents begin to boil. When the vapor reaches the thermometer, the temperature rises to that of the compound with the lowest boiling point. The temperature will stay constant until the first compound has completely boiled away. At which time the temperature will rise to that of the compound with the next lowest boiling point, and so forth until all the compounds of the mixture have boiled off. In this lab you will be driving off the dichloromethane by simple distillation, leaving behind the product in the still pot. Fig. 5-2: Set Up for Fractional Distillation 44 Lab #5: Protocol A. Materials Saturated NaCl Dichloromethane 0.2 M ferric chloride 95% ethanol: water (1:1) methyl acetoacetate centrifuge/centrifuge tubes Organic chemistry kits Heating mantle for round-bottom flasks Vacuum grease Water hoses Ring stands/clamps Balance Small test tubes B. Hazard Communication Avoid getting the dichloromethane on the skin. If this happens, wash with large amounts of soap and water and inform the instructor of the accident. Also avoid deeply breathing the dichloromethane. Do not touch the centrifuge while still spinning. Trying to quickly stop the buckets is dangerous. Furthermore, the pellets are often dislodged during too quick a stop. C. Protocol Part I: Isolation of hydroxy ester product 1. Record the appearance of the yeast mixture and the flask (e.g. Was the parafilm lid still on? Was there a precipitate on the inside of the flask? What was the color of the yeast culture?) 2. Swirl the yeast mixture to distribute the yeast cells. Then pour into 2 centrifuge tubes. Balance the tubes, adding water if needed and place them on opposite sides in the centrifuge. 3. Centrifuge for 10 minutes, then discard the supernatant down the sink. 4. Resuspend the pellet in 50 ml saturated NaCl. 5. Extract twice with 50 ml dichloromethane each time, using the sep funnel. Combine the extracts for simple distillation. Be very gentle during this step to avoid making a mixture of the aqueous and organic phases. You may want to return the extracts to the clean sep funnel at the end and remove the last of the water. Part II: Distillation 1. Obtain a little vacuum grease on a half of a paper towel. Assemble the apparatus for fractional distillation according to the diagram above. At each glass-glass connection take a little bit of vacuum grease and grease the connectors. Hold them together with the plastic clips. The entire apparatus should be clamped to a ring stand. The round-bottom 45 flask will sit in the heating apparatus. The two water hoses are attached to the West condenser. Attach the lower hose to the water source. Place the end of the upper hose in the sink. A clamp or other weight on the hose at the edge of the sink prevents wet papers and wet people. 2. Add the dichloromethane extract by pouring it into the still pot (you may remove the thermometer coupling to pour more easily). Add 3-4 boiling chips to the bottom of the still pot. Have the instructor check your set-up. 3. Turn on the heat to ~6 and turn on the water to the condenser. As the liquid begins boiling, keep an eye on the thermometer and the receiving flask; record the time when the first drop is collected in the receiving flask. 4. Continue boiling until the temperature begins to rise and there is almost no dichloromethane left in the still pot. Then turn off the heat and record the stop time. Part III: Ferric Chloride Test 1. Take about 0.25 ml of the remaining solution at the bottom of the still pot and place in a test tube. In a separate test tube, add 0.25 ml methyl acetoacetate. Add 1 ml ethanol: water (1:1) mix to each tube. 2. Add several drops of 0.2 M ferric chloride and record color changes in each tube. The methyl acetoacetate should turn color (black as a result of forming ferrous chloride) whereas the product (methyl-3- hydroxybutyrate) will not; it will stay yellow. If not all of the starting material was NOT converted to product, then the resulting color will reflect this incomplete conversion (i.e. red or purple or green). 3. Clean up your glassware according to the method described in Lab #1. You may use a final acetone rinse to get rid of the remaining vacuum grease. If you have trouble with the grease, don’t use as much next time. 46 Results and Analysis Part I: 1. What is the purpose of the saturated NaCl? 2. A student has begun a centrifugation step and hears a noise coming from the centrifuge. What is the most likely reason for this noise? What should the student do to prevent this from happening again? Part II: 1. Could you do a simple distillation to separate iodopropane (b.p. 102oC) and water? Why or why not? 2. Why do you think vacuum grease is used with each glass-glass connection? 3. How would your yield be affected if you forgot to turn on the water hoses on the West condenser? Part III: 1. Molecules such as methyl acetoacetate can form enols, where the double bond switches from being between the carbon and oxygen to being between the two carbons. Show how methyl acetoacetate can form an enol. 2. The ferric chloride test is actually a test for enols (i.e. it reacts with the C=C bond). Why would your product be negative? 3. A student noticed that a hole in the lid of his yeast mixture was allowing oxygen to get in for the entire week of incubation. When tested, this sample had a reaction with ferric chloride test indicating that fermentation had not taken place. What had happened? 47 Lab #6: Synthesis of an Ester Fischer Esterification NOTE: THIS LAB WILL BE STARTED BEFORE THE LAB LECTURE TO ALLOW THE MIXTURE TO REFLUX FOR AN HOUR. Introduction: Esters are derivatives of carboxylic acids and are given the generalized formula of RCOOR’. Because esters have no carboxyl hydrogen to donate, they are poor acids. However this means also that esters lack the acrid smell of a carboxylic acid and have a pleasant odor. Many flavorings are esters such as the banana oil that will be prepared tonight. Ethanoic acid (acetic acid) is reacted with the alcohol, 3-methyl-1-butanol, in the presence of a little sulfuric acid. Strong acids are usually used to make esters since it helps in the substitution of the alcohol for the –OH of the carboxylic acid. The reaction occurs with the addition of heat. A reflux condenser is used to condense the vapors and return them to the reaction flask. The ester product is neutralized, dried and weighed. The formation of an ester is an equilibrium process. Therefore, excess acetic acid is used to shift the equilibrium to the right, in favor of products. The final steps of the protocol are directed at neutralizing and removing this excess acid. Introduction to the Reflux Condenser In the kitchen, a lid is put on the pot of boiling food to keep the liquid from boiling away. The reflux condenser is the laboratory equivalent of the lid. It is a double walled tube (usually the Hempel column from fractional distillation) that is placed on top of the heated reaction flask. Water hoses are attached to the reflux condenser so that water flows through the outer jacket. As the reaction flask is heated, the reaction mixture begins to boil. The vapors rise in the reflux condenser until they are cooled by the water jacket back to the liquid which falls back into the reaction flask. For the water to fully surround the reflux condenser, the hoses must be hooked up so that water enters the bottom of the condenser and exits the top. The water pressure should be low to moderate and adjusted so that the vapors are condensing in the bottom third of the condenser. The system is open, as are all heated systems, so there is no stopper or other closure at the top of the reflux condenser. If fumes are ever seen during refluxing, the water pressure should be increased, the heat decreased or both. 48 Lab #6: Protocol A. Materials 3-methyl-1-butanol (isopentyl alcohol) glacial acetic acid concentrated sulfuric acid 5% sodium bicarbonate saturated NaCl anhydrous magnesium sulfate pH paper or red litmus paper organic kits ring stands/clamps vacuum grease heaters boiling chips balances B. Hazard Communication Safety goggles are required for this lab. This lab uses concentrated acids. Contact with the skin can cause burns. If acid accidentally spills on the skin or clothing, wash with large amounts of soap and water. Check frequently during refluxing to make sure there are no vapors released. If fumes are seen and/or smelled, increase the water pressure, decrease the heat or both. Check the heating mantle before use to make sure there are no exposed or damaged wires and that the equipment is dry. Check the glassware before heating for star cracks or chips. Discard all damaged glassware. C. Protocol 1. Pour 15 ml of 3-methyl-1-butanol ( 0.138 mole isopentyl alcohol) and 20 ml glacial acetic acid (0.35 mole) (CAREFUL!!) into a 100-ml round bottom flask. Carefully add 4 ml concentrated sulfuric acid to the contents of the flask, with swirling. Add 2-3 boiling chips to the flask. Place in the heater apparatus. 2. Grease the bottom of a reflux condenser and attach with a metal clamp to the flask. Clamp the condenser to the ring stand to stabilize it. Attach the water hoses. Attach the bottom hose to the water source and put the top hose in the sink. Have the instructor check your set up. 3. Turn on the water and the heat to a setting of 6. Heat for 1 hour, recording both the time the flask began boiling and the total reaction time. 4. Cool and pour the reaction mixture into a sep funnel and add 55 ml water. Rinse the round bottom flask with 10 ml water and add that to the separatory funnel. Stopper the sep funnel and shake it several times with venting. Remove the lower aqueous layer. 49 5. Add 25 ml 5% sodium bicarbonate to the organic layer. Be careful since there is still some acid in the organic layer and it will react with the bicarbonate. DO NOT STOPPER! Swirl gently until there are no more carbon dioxide bubbles released. You may use a glass rod for swirling. 6. When there are no more bubbles, stopper and gently shake the flask, then vent. Repeat until there are no more vapors released when vented. 7. Remove the lower aqueous layer and discard. Add 25 ml 5% bicarbonate to the upper organic layer and repeat steps 5 and 6. Remove the lower layer and test with pH paper (the red litmus paper should turn blue). 8. If not basic, repeat steps 5 and 6. Record how many times you repeated this neutralizing step and how many ml bicarbonate you used. 9. After neutralizing, add 25 ml water to the organic layer. Shake and vent as usual. Add 5 ml saturated NaCl to the sep funnel to help in the phase separation and discard the lower aqueous phase. 10. Place the organic layer in a small beaker. Add 2 g anhydrous magnesium sulfate, swirl and then let sit for 5 minutes. This step removes the water remaining in the organic layer. 11. Decant the ester (organic layer) from the drying agent into a preweighed beaker. Weigh the product. 12. Calculate the percent yield as: __mole actual yield__ x 100 mole theor. yield For the theoretical yield, decide which reagent is the limited reagent. (In this lab, the mole of each reagent is given in step 1.) Then look at the equation and compare the mole of limiting reagent with the mole product. In this case there is a 1:1 molar ratio between limiting reagent, so the theoretical yield is the mole of limiting reagent. All the mole of limiting reagent get converted to product. To determine the actual yield divide the gram product from step 11 by the FW of the isopentyl acetate, C7H14O2. (Add the atomic weights of each atom to get FW; given in g/mole.) Results and Analysis 1. Aspirin (acetylsalicylic acid) plus methanol in acid makes an ester that is known as oil of wintergreen. Draw the ester. 2. What is your theoretical yield? List 4 places where you have lost product. 3. Define the term “limiting reagent” as it was used above. 50 Lab # 7: Protein Determination Introduction Laboratories often use assays to quantify compounds for a number of reasons; 1) to determine the effectiveness of each step of a purification procedure, 2) to determine the purity of a product, and 3) to determine the concentration of an analyte. Protein assays are normally done on liquid samples but there are procedures for determining protein concentration in gels and other types of mediums. In this laboratory exercise you will use one of the more common instruments used to assay protein concentration in a liquid sample, a UV/VIS (ultraviolet/visible) spectrophotometer. A UV/VIS spectrophotometer is an instrument that passes light of a given wavelength through a liquid. Molecules that contain double or triple bonds will usually absorb the light at a characteristic wavelength of between 200 and 750 nm Each molecule has a wavelength at which it absorbs the most light so in actuality you can determine the concentration of a sample just by measuring its absorption at a specific wavelength depending on its absorption characteristics. The colored product produced in this laboratory exercise absorbs the maximum amount of light at 595nm, and the more product present, the more light is absorbed. This may be read off the spectrophotometer in a log scale called Absorbance units, or Abs. Once the Absorbance or color intensity has been measured, it must be compared to a standard curve. The standard curve is created from a series of known amounts of material to be assayed that is treated in the same way as the sample. The Absorbance units (y-axis) are plotted against the concentration (x-axis) to get a straight line relating concentration to amount of color. Then the sample is compared to this standard curve so that the concentration of substrate in the sample may be determined. Beer’s Law or A=mC or A =(Eb)C The line on a calibration curve has an equation Y=mX +a where m = slope and a = the Y intercept; in this case the Y intercept equals 0. Thus the equation for the line is simply A=mC or absorbance = (slope of calibration line)(concentration of analyte). Two parameters are known to affect the steepness of the slope. 1) The chemical and physical nature of the analyte or material being measured, which is going to be specific for each pure analyte. This inherent tendency of a material or analyte to absorb light at a particular wavelength is called its absorptivity. The name given to this constant is absorptivity constant or extinction coefficient also known as E. 2) The path length of the sample, which is usually 1 cm since cuvettes are usually 1cm wide. It is designated as b. More information on this equation and E is given later and also in chapter 20 in the book, Basic Laboratory Methods for Biotechnology. Good laboratory practice requires that the standard curve and the sample assays are performed using use the same reagents. This eliminates error due to variations in the assay from day to day. In the first part of this lab, a stock bovine serum albumin (BSA) solution is prepared and its actual concentration determined. Since it is being used for the standard 51 curve, it is important that its actual concentration is known. In the second part of the lab, it is diluted by serial dilution to make the protein standard curve. The unknown samples will also be diluted to hopefully bring their protein concentrations within the range of the assay. Then the protein color reagent will be added to all tubes. After an appropriate incubation time, the resulting color is read in the spectrophotometer. Determination of Protein Concentration in a Solution Four spectroscopic methods are routinely used to determine the concentration of protein in a solution. These include measurement of the protein's intrinsic UV absorbance (1) and three methods, which generate a protein-dependent color change; the Lowry assay (2), the Smith copper/bicinchoninic assay (3) and the Bradford dye assay (4). Although one or more of these methods are used routinely in almost every biochemical laboratory, none of the procedures are particularly convenient, for the reasons described below. The first, UV absorbance, requires that a pure protein with known extinction coefficient, otherwise known as an absorptivity constant, be used, in a solution free of interfering (UV absorbing) substances. For example, a pure solution of bovine serum albumin (BSA) has an E = 0.667 mg cm-1 ml so that at A280 a 1mg/ml solution in a 1 cm pathlength cuvette has an absorption of 0.667. If the extinction coefficient is unknown, as an approximation, the protein concentration of a solution can be estimated by using either of the following equations: A280 = 1 A (ml/cm mg) x [Conc.] (mg/ml) x 1 (cm) A205 = 31 A (ml/cm mg) x [Conc.] (mg/ml) x 1 (cm) Different proteins, however, have widely different extinction coefficients at both 280 and 205 nm, and therefore concentration estimates obtained this way should be viewed with considerable skepticism. Again, this assay requires that the protein solution be free of other UV absorbing substances, and that the measurements be made using the appropriate cuvette. The Lowry and copper/bicinchoninic assays are based on reduction of Cu 2+ to Cu 1+ by amides. Although this makes them potentially quite accurate, they require the preparation of several reagent solutions, which must be carefully measured and mixed during the assay. This is followed by lengthy, precisely timed incubations at closely controlled, elevated temperatures, and then immediate absorbance measurements of the unstable solutions. Other substances frequently present in biochemical solutions, including detergents, lipids, buffers and reducing agents, may affect both assays. This requires that the assays also include a series of standard solutions, each with a different, known concentration of protein, but otherwise having the same composition as the sample solutions. The Bradford dye assay is based on the equilibrium between three forms of Coomassie Blue G dye. Under strongly acid conditions, the dye is most stable as a doubly-protonated red form. Upon binding to protein, however, it is most stable as an unprotonated, blue form. 52 Red <=> (470 nm) Green <=> (650 nm) Blue <=> (590 nm) Blue-Protein (590 nm) H+ H+ The Bradford assay is faster, involves fewer mixing steps, does not require heating, and gives a more stable colorimetric response than the assays described above. Like the other assays, however, its response is prone to influence from non-protein sources, particularly detergents, and becomes progressively more nonlinear at the high end of its useful protein concentration range. The response is also protein dependent, and varies with the composition of the protein. These limitations make protein standard solutions necessary. 53 Lab #7: Protocol A. Materials Bradford reagent or prepared Coomassie Blue Reagent (described below in appendix) Reagent grade bovine serum albumin (BSA) 1 ml test tubes volumetric flask vortex mixer analytical balance parafilm spatula UV Spectrophotometer/cuvettes Two unknown protein samples B. Hazard Communication Make sure that the wiring for the spectrophotometer is not frayed and that the instrument is not in or near water. C: Protocol Part I: Determining the concentration of BSA 1. Turn on the spectrophotometer and let warm up about 15 minutes. Set wavelength to 280 nm. 2. Prepare the 2mg/ml solution of BSA. It is suggested that you prepare enough solution so that you can accurately weigh out the BSA using the analytical balance and a volumetric flask (e.g. 10 ml flask). 3. Determine the absorbance of the solution. 4. Calculate the corrected concentration of the solution using Beer’s Law and the given E value for BSA. (A=EbC) Part II: Your instructor will tell you if you are doing a standard or a micro assay. Determining the concentration of an unknown protein sample Standard Protein Assay Procedure (200 - 2000 µg/ml protein): Prepare six standard solutions (1 ml each) containing 0, 250, 500, 1000, 1500 and 2000 µg/ml BSA. Following the directions provided by the Bio-Rad prepare the reagent and set up the protein assay in test tubes. To mix the samples, cover with parafilm and gently invert several times to mix or vortex. Examine the absorption readings of the standards and samples. If there are any readings greater than 2, or if any sample has an absorbance greater than the greatest absorbance for any of the standards, dilute the sample by a known amount and repeat the assay. Microassay Procedure (<50 µg/ml protein): Prepare five standard solutions (1 ml each) containing 0, 10, 20, 30, 40 and 50 µg/ml BSA. Follow the procedure described above for the standard assay procedure. 54 Results and Analysis The Bradford assay gives a hyperbolic plot for absorbance versus protein concentration, but within a range of relatively low protein concentrations, the hyperbolic curve can be approximated reasonably well by a straight line. To determine the protein concentration of a sample from it absorbance, prepare a standard curve as described below to find the concentration of standard that would have the same absorbance as the sample. Using the graphing function of your computer or Excel, graph Absorbance. on the Y-axis and ug/ml BSA on the X-axis. Examine the graphed points and decide if any should be rejected. (Often a single point can be rejected without invalidating the standard curve, but if more than one point appears questionable the assay should be repeated). Review chapter 10 on graphing in the book, Basic Laboratory Methods for Biotechnology, if you do not remember linear regression analysis. Ask your instructor for help with Excel if you do not remember how to plot using a trendline or starting the line at X=0, Y=0. Appendix Dye stock - Coomassie Blue G (C.I.# 42655) (100 mg) is dissolved in 50 ml of methanol. (If turbid, the solution is treated with Norit (100 mg) and filtered through a glass-fiber filter.) The solution is added to 100 ml of 85% H3PO4, and diluted to 200 ml with water. The solution should be dark red, and have a pH of -0.01. The final reagent concentrations are 0.5 mg/ml Coomassie Blue G, 25% methanol, and 42.5% H 3PO4. The solution is stable indefinitely in a dark bottle at 4°C. Assay reagent - The assay reagent is prepared by diluting 1 volume of the dye stock with 4 volumes of distilled H2O. The solution should appear brown, and have a pH of 1.1. It is stable for weeks in a dark bottle at 4°C. 55 Lab #8: Preparation of Yeast Media Introduction: Media is the term used for the growth solution for cells grown in the lab. Media may be either chemically defined or complex. Chemically defined media contain only substances that are clearly identified. One type of chemically defined media is minimal media. This is media that contains the minimal ingredients to support life. There are different minimal media for bacteria, yeast, animal or plant cells. For example, the minimal media for growing human cells would have to contain vitamin C since human cells are unable to make this essential nutrient and must obtain it from their food. (See Lab #8). Rich or complex media contain a variety of nutrients and some ingredients that are either unidentified or might be present in variable concentrations. It is impossible to completely define rich media. Yeast extract, the extract of all the water-soluble components of yeast, is sometimes added to media for growing a variety of microorganisms. The yeast extract cannot be chemically defined and its composition will vary depending on the growing conditions of the source yeast. Selective and differential media may be either chemically defined or rich. Selective media acts by killing microbes that lack a specific phenotype. For example, bacteria media containing the antibiotic penicillin will support the growth only of penicillin-resistant bacteria. Therefore the media is selecting for a specific trait. Differential media will distinguish between microbes that possess a given trait and those that do not, often by color or shape of the colony. In this lab, we will prepare a yeast minimal media (Y-min), a yeast rich media (YPD) and a differential media for yeast. The differential media will contain bromthymol blue which turns color if the yeast is able to use galactose to ferment. The media will be sterilized and then poured onto plates. Next week, the plates will be streaked with a yeast strain, YPH 274. Sterilizing Media Media is sterilized in order to avoid the growth of undesirable microorganisms. There are two major sterilization methods in common use in the lab: autoclaving and filter sterilization. An autoclave is in effect, a large pressure cooker, using steam and pressure to kill all undesirable microorganisms. Typically 15 to 20 minutes autoclaving at 15 lb/in2 pressure is sufficient to sterilize heat-resistant media and glassware or for discarding biological waste. However heavily contaminated material such as rabbit pellets require 180 minutes of autoclaving at 15 lb pressure. Some media ingredients are sensitive to heat and will break down in the autoclave. These ingredients are sterilized in the lab by filter sterilization. A sterile filter with holes or pores of 0.22 M is used to filter out bacteria and other contaminating cells. Filters with a pore size as small as 0.01 M will prevent passage of viruses and protein. Small amounts of media are filter sterilized with a syringe filter. This is a filter that is in a sterile casing adapted to fit at the end of a syringe. The media is loaded into the syringe, the filter is attached and the apparatus placed on top of a sterile test tube, and 56 pushing the plunger of the syringe forces the media through the filter and into the test tube. You can sterilize more media with the same filter but it must be removed from the syringe when pulling out the syringe barrel otherwise material on the bottom of the filter will be drawn up through the filter destroying the integrity of the filter. Larger amounts of media are filtered with a larger apparatus, which are usually a self-contained unit with chambers above and below the filter. The unsterile media is placed in the upper chamber. The lower chamber is fitted with a side arm that is attached to a vacuum so that the media is pulled through the filter by the vacuum in the lower chamber. To maintain sterility, it is best to filter sterilize media and streak the plates in the appropriate hood. Fig 9.1: Filter Sterilization 57 Lab #8: Protocol A. Materials Part I: Yeast Minimal Media Bacto-yeast nitrogen base without amino acids Glucose Bacto-agar Erlenmeyer flasks Stir bars/stirrers Aluminum foil Part II: Yeast Rich Media Bacto-peptone or tryptone Glucose Bacto-agar Bottles Stir bars/stirrers Part III: Yeast Differential Media Bacto-yeast extract Bacto-peptone or tryptone Bacto-agar Bromthymol blue solution (4 mg/ml stock solution) galactose Bottles Stir bars/stirrers Sterile filters and syringes Part IV: Pouring the Plates Sterile petri dishes Next Week: Yeast strain YPH274 inoculating loops plates from last week bacto-incinerators NOTE: YOU WILL BE ASSIGNED TO MAKE EITHER THE MINIMAL MEDIA (PART I) OR THE RICH MEDIA (PART II) TO BE SHARED WITH THE OTHERS. EACH GROUP WILL MAKE THE DIFFERENTIAL MEDIA (PART III) FOR THEMSELVES. B. Protocol Part I: Preparation and Use of the Hood 1. Start with a completely clear surface 2. Swab the surface liberally with 70% alcohol 3. Bring onto the surface only those items you will need and arrange them so you have access to all items without having to reach over one to get to another. Swab bottles, pipetters, containers, and the plastic bags covering the plates and plates etc. with 70% alcohol. 4. Let the hood and items dry before using them. 5. Work within your range of vision 6. Mop up any spillage immediately and swab the area with the 70% alcohol 7. Remove everything when you have finished, and swab the work surface down again 58 Part II: Preparation of Yeast Minimal Media (Y-min) 1. Calculate the amount of each ingredient needed to make 200 ml of this chemically defined media. Remember that “%” means g/100 ml water. Y-min media 0.67% bacto-yeast nitrogen base without amino acids 2% glucose 2 % bacto-agar 2. Weigh out these ingredients, recording the weights as usual. Dissolve the first two ingredients in about 100 ml water by swirling the flask. . Pour into an Erlenmeyer flask containing the agar and cover with aluminum foil and autoclave tape, BTV. Volume measurement using a flask can be off + or – 5% but that error is acceptable for this experiment. Label the tape and put in the autoclave. Part III: Preparation of Yeast Rich Media (YPD) 1. Calculate the amount of each ingredient needed to make 200 ml of this rich media. Remember that “%” means g/100 ml water. YPD media 2% bacto-peptone 2% glucose 2% bacto-agar 2. Weigh out the first two ingredients, recording the weights as usual. Dissolve in about 100 ml water. Pour into an Erlenmeyer flask containing the agar, BTV, and cover with aluminum foil. Label and put in the autoclave. Part IV: Preparation of Yeast Galactose Indicator Media 1. Calculate the amount of each ingredient needed to make 100 ml of this differential media. Galactose indicator media: 1% bacto-yeast extract 2% peptone 2% bacto-agar 2 ml bromthymol blue solution (filter sterilize) 10 ml 10% galactose (filter sterilize) 2. Weigh out these ingredients, recording the weights as usual. Dissolve the first three ingredients in about 50 ml water. Pour into a flask containing the agar, bring up to approximately 90 ml with water, and cover with foil. Label and put in the autoclave. 3. When all bottles are in the autoclave, turn on the autoclave and autoclave for 20 min at 15 lb pressure (121oC). 4. While the media is autoclaving, prepare 10 ml of 10% galactose. 5. In the hood, remove the plunger from a 10 ml syringe. Wipe down the outside of the syringe barrel with alcohol. Remove the tip to the syringe barrel and discard. Open up the sterile filter apparatus and attach to the bottom of the syringe. Place into the top of a sterile test tube. Pour the galactose into the syringe and push through the filter into the sterile tube with the plunger. Replace the cap on the sterile tube with the now sterile galactose. Discard the filter. 6. Repeat the filter sterilization step with the bromthymol blue solution. 59 7. When the sterile galactose indicator media is cool enough to handle, pour the sterile galactose into the media and add 2 ml of the sterile bromthymol blue solution to the media. Note, there are several ways you could prepare this media; this is one way. You could also filter-sterilize the correct volume of galactose solution directly into the media. You could add the agar to the initial solution, heat it to dissolve the agar and then BTV. When you’re working in a lab, you’ll develop your own routine based on lab input or follow established procedures that work best for that lab. Part V: Pouring the Plates (Your instructor will demo this technique) Pouring the plates must be done using sterile technique so as not to produce contamination. 1. Set up a stack of either two or three plates, depending upon how many plates you feel you can successfully hold at one time. Take a sterile Y-min bottle and record the label information. Remove the top to the bottle with your pinkie finger and palm of the hand you will be pouring with. If you cannot manipulate the bottle and hold the cap, put the cap down on the surface with the open side down. Then pour the media into the bottom plate of the stack until the media is just covers the bottom of the plate, swirl gently. Pour the middle plate of the stack, swirl. Repeat for the top plate. Replace the top on the bottle. 2. After you have poured at least two plates, let another student practice pouring plates. 3. Repeat procedure with the rich media and the galactose indicator media. 4. Allow plates to dry and package according to instructor’s directions. NEXT WEEK: Part V: Streaking the Plates with Yeast 1 Prepare the hood for use. Place your plates from last week right side up in the hood. Obtain a sterile culture of YPH274 yeast and an inoculating loop. 2. Sterilize the loop by holding it in the incinerator until it glows red. Cool by stabbing it into the corner of the agar of a Y-min plate. (Lift the lid slightly to allow enough space for putting in the loop but not so much that you uncover the dish and allow air-borne particles to fall onto the plate.) 3. Remove the top of the yeast tube and obtain a loopful of yeast. Place it on the plate near the side. 4. Sterilize the loop, cool it and drag it through the drop of culture, moving back and forth in a zigzag pattern. Turn the plate 90o, flame the loop, cool it and repeat. Turn the plate another 90o, flame the loop, cool it and repeat. (See Fig 11-1). 5. Continue with the other 2 Y-min plates and the rich media and galactose indicator plates. 6. Incubate inverted (upside down) for at least 48 hours at 30oC. Record your results. 60 Fig. 9.2: Three Phase Streaking of Plates 61 C. Results and Analysis 1. Would you expect to see more growth on the rich media plates as compared to the minimal media plates? Would you expect to see faster growth on the rich plates? Why or why not? 2. Are the YPH274 yeast able to use galactose for fermentation? Why did you answer as you did? 3. Why was the galactose for the galactose indicator media filter sterilized instead of autoclaved? 4. A student makes some rich media plates and streaks them with a yeast strain. Upon examination of the plates, the student finds colonies of different sizes and colors. Give at least 2 ways the student could have introduced contamination. 62 Lab #9: Acid Phosphatase Kinetics and Inhibitors Introduction: Enzymes are proteins that catalyze reactions in the cell. Virtually all chemical reactions of living organisms are mediated by enzymes. Therefore their characterization is crucial for the understanding of biology. The tertiary folding of the enzymes creates a characteristic pocket or active site into which the substrate or substrates exactly fits. This allows for the reaction to occur without using up the enzyme—hence it is a catalyst. The rate at which an enzyme catalyzes a reaction is characteristic for that enzyme at a given pH, temperature and ionic strength. Therefore, rate kinetic studies are often performed on enzymes. The rate, usually given as Km, is related to the rates at which the enzyme-substrate complex is formed and the rate at which product is made. Thus k1 k3 Enzyme + Substrate Enzyme-Substrate complex Enzyme + Product k2 k4 The Michaelis rate for the enzyme, Km, is defined as (k2 + k3)/k1 and it has units of concentration. In fact, Km is defined as the substrate concentration at which the enzyme catalyzes ½ the maximal velocity for the reaction. Note, k2 and k3 are involve in dissociation of the enzyme-substrate and k1 is involved in formation of the complex. It doesn’t include k4 because conversion of product to substrate cannot occur or does not occur readily (i.e. it would take a large amount of product to shift the equilibrium to formation of substrate). The question becomes how to determine this Km. Michaelis and Menten found that they could measure the velocity of the reaction at different concentrations of substrate. Their equation for the reaction was Vo = __(Vmax) [S]__ where Vo is velocity, [S] is the substrate concentration Km [S] and Vmax is the maximum velocity of the enzyme. Vmax is reached when all of the enzyme is bound to substrate. Plotting Vo (y-axis) vs [S] (x-axis) produces the following graph. Vo [S] 63 Lineweaver and Burke rearranged the Michaelis-Menten equation to a more useful form: _1_ = _Km_ . __1__ + __1__ Vo Vmax [S] Vmax This is identical to the plot of a straight line: Y = MX + B. In the Lineweaver-Burke equation Y = 1/Vo; X = 1/[S]; the slope, M = Km/Vmax and the Y-intercept, B = 1/Vmax (The X-intercept is –1/Km.) Therefore, an experiment may be performed in which the velocity of a reaction (in this lab measured as the production of color which may be read in a spectrophotometer) is determined at different concentrations of substrate. By using the Lineweaver-Burke equation to plot the results, the Km for the enzyme may be determined. A typical Lineweaver-Burke plot of an enzymatic reaction is given in Fig. 12-1. 1/Vo slope = Km/Vmax 1/Vmax - 1/Km 1/[S[ Fig 10-1: A Lineweaver-Burke plot Either competitive or noncompetitive inhibitors may inhibit the activity of enzymes. Competitive inhibitors are molecules that sit in the active site of the enzyme and prevent the entry of the substrate. Competitive inhibitors frequently look similar to the substrate. A Lineweaver-Burke plot of a competitive inhibitor will have the same Yintercept as that of the substrate. 64 Noncompetitive inhibitors do not sit in the active site but will attach to an allosteric site and change the shape of the enzyme and therefore the shape of the active site. This results in preventing the substrate from binding to the enzyme and being converted into product. In a Lineweaver-Burke plot of a noncompetitive inhibitor, the Xintercept, -1/Km, is the same as that for the substrate. Thus the two types of inhibitors may be distinguished from each other. [I] Competitive Inhibitor [I] Noncompetitive Inhibitor Fig 10-2: Competitive and Noncompetitive Inhibitors Value of Knowing Km and Vmax for an Enzyme It is fairly obivious that understanding how an inhibitor works is important, especially in the pharmaceutical industry; but what can Km and Vmax tell you about an enzyme? For most enzymes Km lies between 10-1 and 10-7 M and depends on the particular substrate and on environmental conditions such as pH, temperature, and ionic strength. Since Km is the concentration of substrate at which half the active sites are filled it is the concentration required for significant catalysis to occur. In fact, experimental evidence indicates that Km provides an approximation of substrate concentrations in vivo. Km is also related to the rate constants of the individual steps in the catalytic scheme (i.e. formation and dissociation of the enzyme-substrate complex). Under conditions where the dissociation of ES is much greater than the formation of P, Km is a measure of the strength of the ES complex. The maximal rate Vmax, reveals the turnover number of an enzyme, which is the number of substrate molecules converted into product by an enzyme molecule in a unit time when the enzyme is fully saturated with substrate. The turnover numbers of most enzymes with their physiological substrates fall in the range of 1 to 104 per second. 65 The purpose of this experiment is to determine the Vmax and Km values for wheat germ phosphatase. Phosphatase is an enzyme that removes a phosphate group from a molecule. The substrate is p-nitrophenyl phosphate. The velocity of the reaction is monitored by measuring the absorbance of the colored product at 405 nm in a spectrophotometer. In Part III, two inhibitors of the reaction, sodium phosphate and sodium fluoride are used. One is a competitive inhibitor and the other is a noncompetitive inhibitor. 66 Lab #9: Protocol A. Materials AT Buffer (0.01 M sodium acetate/acetic acid buffer, pH 5 with 0.01% Triton X-100; 1 l for 4 groups) wheat germ acid phosphatase (20 mg enzyme at 0.5 U/mg dissolved in 10 ml AT buffer; stored on ice. Enough for at least 24 students.) p-nitrophenyl phosphate (100X stock = 250 mg/6.7 ml; FW = 371.1 g/mole) 1 M KOH 100X stock sodium phosphate, dibasic (0.355 g in 25 ml AT buffer; FW = 142.0 g/mole) 100 X stock sodium fluoride (0.32 g in 30 ml AT buffer; FW = 42.0 g/mole) 50-ml graduated cylinders micropipettes 5-ml and 10-ml pipettes and bulbs test tubes spectrophotometer cuvettes timers B. Hazard Communication The KOH and the sodium phosphate are caustic. Treat with caution. C. Protocol Part I: Determination of Km 1. Label eight tubes 1-8. 2. Measure out 150 ml of AT buffer. Add 1.5 ml 100X stock p-nitrophenyl phosphate. Label as “AT+S buffer”. The substrate concentration of this buffer is 1.0 mM. 3. Calculate the amount of substrate you will need for 3.0 ml of the samples in tubes 2-8. Enter these values on the table on the next page. Calculate the ml AT buffer will be needed to BTV to 3.0 ml in tubes 2-8. Enter these values on the table on the next page. 4. Make up tubes 1-8 according to your calculations. 5. Set a timer and at 0 seconds, add 50 l enzyme to tube #1. Exactly 30 seconds later, add 50ul enzyme to tube #2. Continue at 30-second intervals until the enzyme has been added to each tube. 6. At exactly 5 minutes add 250 l 1 M KOH to tube #1. Repeat at 30 second intervals for each of the other tubes. Each tube should have the same volume and should have incubated with the enzyme exactly 5 minutes before the reaction was stopped with KOH. 7. Read the Absorbance at 405 nm and record on the table above. 67 # Substrate conc (mM) 1 0 (blank) 2 0.01 mM 3 0.05 mM 4 0.1 mM 5 0.25 mM 6 0.5 mM 7 0.75 mM 8 1.0 mM Table I: Determination of Km ml AT ml AT+S buffer buffer Abs405 3.0 ml 0.0 ml 0.0 ml 3.0 ml 0 1/[S] ----- -1/Abs ----- 68 Part II: Inhibitors of Acid Phosphatase 1. Sodium phosphate is an inhibitor of the enzyme. Begin by placing 0.3 ml 100X sodium phosphate stock into 29.7 ml AT buffer to make 1.0 mM Na2HPO4 (Sodium phosphate, dibasic). Label as “AT+P buffer”. 2. Place 0.3 ml 100 X sodium phosphate stock into 29.7 ml of the AT+S buffer. Label as “AT+S+P buffer”. 3. Label 8 tubes 1-8. 4. Calculate the dilutions of AT+P and AT+S+P buffers as for Part I and record on the table. 5. Once the substrate-inhibitor tubes are ready, begin the experiment as before by adding 50 L enzyme to each tube at 30 second intervals. Quench with 1 M KOH at 5 minutes. Read Absorbance and record on the table. # Table 2: Determination of Km with Sodium Phosphate Inhibitor Substrate ml AT+P ml AT+S+P conc buffer buffer Abs405 1/[S] 1/Abs (mM) 1 0 (blank) 2 0.01 mM 3 0.05 mM 4 0.1 mM 5 0.25 mM 6 0.5 mM 7 0.75 mM 8 1.0 mM 3.0 ml 0.0 ml 0.0 ml 3.0 ml 0 69 6. Sodium fluoride is another inhibitor of the enzyme. To begin, place 0.3 ml 100X NaF stock into 29.7 ml AT buffer. This makes a 2.5 mM NaF solution. Label as “AT+F buffer”. 7. Place 0.3 ml 100 X NaF stock into 29.7 ml of the AT+S buffer. . Label as “AT+S+F” buffer. 8. Set up and do the experiment as before. Determination of Km with Fluoride Inhibitor # Substrate conc (mM) ml AT+F buffer ml AT+S+F buffer 1 0 (blank) 3.0 ml 0.0 ml 2 0.01 mM 3 0.05 mM 4 0.1 mM 5 0.25 mM 6 0.5 mM 7 0.75 mM 8 1.0 mM 0.0 ml 3.0 ml Abs405 1/[S] 1/Abs 0 Part III: Data Analysis 1. For each set of data, calculate the inverse of the substrate concentration (1/[S]) and the negative inverse of the Absorbance (1/Abs) Record on the tables. 2. Plot these values on a single graph, putting 1/[S] on the X-axis and 1/Abs on the Yaxis. Connect the values using linear regression analysis on the computer as before. 3. Calculate the Vmax and the Km for the enzyme and the enzyme in the presence of each of the inhibitors. 70 D. Results and Analysis 1. Which of the inhibitors is a competitive inhibitor and which is a noncompetitive inhibitor? Why did you answer as you did? 2. How well did your data fit a straight line? Think of two things you could do to get cleaner data. 3. What would be the Km if twice the enzyme were used? Explain. 71 References Lab #1: Hadfield, Jillian, Laboratory Solutions, in press. (Ch 24) Lab #2: Ibid. (Ch. 21) Lab #3: from Sonia Wallman, New Hampshire Technical College Lab #4: Roberts, Royston M, John C. Gilbert and Stephen F. Martin, Experimental Organic Chemistry, A Miniscale Approach, Harcourt Brace, 1994.(Ch. 5) Lab #5: Ibid (Ch 17) Lab #6: from Sackett, Deborah, Austin Community College. Lab #7: Stewart, Kent K. CH 204: Introdcution to Chemiscal Principles for Life Sciences, UT Duplicating Services, 1999 (Laboratory 10) Lab #8: Boyer, Rodney F., Modern Experimental Biochemistry, 2e, Benjamin/Cummings, 1993. (Experiment 15) Lab #9: from Barreto Jose, Florida Gulf Coast University and from Roberts, et al. (Ch. 25) Lab #10: From Lisa Seidman, Madison Technical College Lab #11: Porter, Sandra G., Bio 282 Media Preparation, and Madison Technical College, 1996 (Ch 8) Lab #12: Minch, Michael J., Experiments in Biochemistry, Prentice-Hall, 1989. (Ch 17) 72 Appendix A: Mandatory laboratory safety rules Modern laboratories are equipped with supplies and equipment that may pose a hazard if used carelessly. Following safety rules, paying attention to what you are doing, and using common sense is the best way to make your experience in this course a safe one. Health and safety are paramount values in science classrooms, laboratories and field activities. You are expected to learn, understand and comply with ACC environmental, health and safety procedures and agree to follow the ACC science safety policy. You are expected to conduct yourself professionally with respect and courtesy to all. Anyone who thoughtlessly or intentionally jeopardizes the health or safety of another individual will be immediately dismissed from the day’s activity, may be withdrawn from the class, and/or prohibited from participating in future activities. Specific safety training will take place before most activities. If you are late and miss this training, you will not be able to participate in the activity. You can read the complete ACC science safety policy at http://www2.austincc.edu/sci_safe/ General laboratory safety rules 1. Never eat or drink in the lab. Do not bring food or drinks into the lab. 2. Avoid wearing contact lenses in the laboratory. Chemical vapors can permeate lenses or become trapped behind them, potentially damaging your lenses or eyes. 3. Do not come into contact with another’s body fluids (blood, saliva, urine). Assume all body fluids are infectious. 4. Wear UV-rated safety glasses or goggles and gloves at all times while in the lab. Change your gloves often, and while wearing gloves, do not touch door handles, water taps, computers, telephones or other objects that may be touched by people not wearing gloves. Gloves will be provided, but students should provide their own UVrated safety glasses. 5. Remove your gloves and wash your hands immediately after handling animals or body fluids. 6. Keep book bags and other personal items off the tables and floor during lab. You can store your personal items in the labeled cupboards or on empty chairs so that they are out of the way. 7. Do not wear loose or flowing clothing or dangling jewelry in the laboratory. Pin up long hair or confine it under a hat. Some of the substances used in the lab may stain or damage clothing. You may wish to wear a lab coat or apron for protection, or wear clothing that you don’t mind being stained or damaged. 8. Do not wear sandals or open-toed shoes. Flat-heeled, full-coverage shoes of leather or other impermeable material are best. 9. Report broken glass immediately to your instructor, and always dispose of broken glass in its designated container. Do not place non-glass items in these containers. 10. Clean up your work stations, wipe your lab bench and wash your hands before leaving the lab room. 73 General safety procedures 1. Each student should know the location and proper use of each of the following: gloves, goggles, safety shower, eyewash station, biohazard container, broken glass disposal box, fire extinguisher, first aid kit, and hazardous material spill cleanup kit. 2. If you have any doubt about a proper safety procedure in a specific instance, ask your instructor. Handling chemicals 1. Wear gloves and goggles when handling potentially hazardous chemicals. 2. Read the label completely before opening a chemical bottle (pay special attention to warning labels). 3. Open volatile organic solvents only in a fume hood. 4. Close all containers immediately after using. 5. Always handle chemicals with care to avoid spills. Clean up and dispose of if not hazardous. 6. Report any spills of a potentially hazardous chemical immediately to your instructor. 7. Follow your instructor’s directions on how and when to mix chemicals. 8. Don’t taste chemicals or smell fumes directly. 9. Don’t use your mouth to pull liquid into a pipette. 10. Always use clean glassware to prevent contamination. 11. Don’t pour unused chemicals back into storage containers where it may contaminate the rest of the reagent. Dispose of unused chemicals in proper waste containers. 12. Do not flush chemicals or cleanup materials down the drain without instructor’s consent. 13. Consult with your doctor about any special health conditions that you may have, such as asthma, allergies, or pregnancy. 14. Clean up work areas thoroughly when you are finished. Always clean up shared areas such as balances and stir plates. Never leave spilled chemicals sitting on a balance, even if you did not spill it. They can corrode the instrument. 15. Wash hands prior to leaving the laboratory. Handling biohazards 1. When working with microbes, always assume they are infectious. Avoid touching contaminated objects to any other objects, even the floor and counters, and avoid direct contact, especially around broken skin. 2. Always wear gloves and goggles when working with microbes or body fluids. 3. Dispose of all potentially contaminated objects in a biohazard bag or a container filled with a 10% bleach solution, or follow your instructor’s directions. 4. Spray and wipe the work areas with 10% chlorine bleach solution before and after lab. 5. Wash hands immediately after handling a biohazard. 74 Handling mechanical hazards 1. 2. 3. 4. Never touch a rapidly moving machine, such as a centrifuge, while it is moving. Distribute weights evenly in a centrifuge to prevent vibrations and breakage. Do not leave a running centrifuge unsupervised. If the centrifuge is vibrating excessively or “walking” across the tabletop, turn it off immediately. Handling electrical hazards 1. Do not use equipment that has any frayed or damaged wiring or plugs. Report any uninsulated wires to your instructor. 2. Always make sure the area around all electrically powered equipment is dry before turning on the power. 3. Gel electrophoresis poses a high risk for electrocution. When assembling or disassembling the gel apparatus, always be sure that it is unplugged. Connect the power supply and turn on the power supply only under the supervision of your instructor. Handling glassware 1. Dispose of disposable glass items such as capillary tubes and cover slips in a hardsided box labeled “Glass Disposal”. 2. Do not use broken or cracked glassware. If you break a glass item, report the incident to your instructor and dispose of it in the “Glass Disposal” box. 3. NEVER put broken glass or disposable glass items with sharp edges in the ordinary trash can. This poses a serious hazard to the person who must empty the trash. 4. Avoid rapid temperature changes of any glassware, as this will often cause the glass to break. Do not place a cool glass container on a hot surface (such as a hot plate), and do not place a hot glass container on a cold surface or in a cold environment (such as a refrigerator). 5. Report any cuts immediately to your instructor, and wash the wound thoroughly in running water. Check for glass in the wound, remove if necessary, dry the skin, and apply a bandage. 6. Do not shake glass thermometers, and lay thermometers away from the edge of a bench on a towel or screen to avoid dropping it on the floor. If a thermometer breaks, immediately inform your instructor. Handling Bunsen burners 1. 2. 3. 4. 5. 6. Check gas hose for cracks every time before using. Make sure hose fits securely. Make sure the striker produces sparks before turning on the gas. Stand back, open gas, use striker, and adjust flame. Flame should be blue. Adjust oxygen intake if you have a yellow flame. If the flame sputters or goes out, immediately turn off gas and inform instructor. 75 7. If you smell gas, turn off gas and immediately inform instructor. 8. If you are going to heat a glass container, check container closely for cracks or stars. Do not use glass that is cracked or starred. 9. Glass containers must have an opening for vapors and heat to escape when heated. 10. Use beaker tongs, hot hands, insulated gloves or test tube holders when handling hot glass. 11. Remember: hot metal and glassware can look just like cool metal and glassware. Be aware of hot materials and make sure no one handles them. Don’t leave a hot object unattended without notifying every person in the room. 12. When heating test tubes over a flame, move the tube back and forth at an angle to avoid splattering, and point it away from others and yourself to avoid being burned by splattered liquids. Disposal procedures 1. Treat all biological and chemical materials as if it were hazardous waste, unless notified otherwise by your instructor. 2. Do not pour chemicals, solutions, or biologicals down the drain without permission by your instructor. 3. Dispose of wastes in the proper labeled waste containers, as indicated by your instructor. Chemicals should be poured by funnel into a labeled chemical waste bottle. Biohazards should be placed into a biohazard bag (do not overfill), sealed with autoclave tape, and autoclaved until sterile. Glass should go into a labeled glass disposal box. Accident procedures 1. Try to contain any spills without endangering yourself and others. Spill socks and pillows, or paper towels if necessary, can be used to contain a spill and keep it from spreading. Notify the instructor immediately when a spill has occurred. 2. If a caustic chemical is splashed into your eyes, notify the instructor or another student immediately so that you can be assisted to the nearest eyewash station as quickly as possible. Continue to wash your eyes for at least 20 minutes while emergency personnel are being called. 3. If caustic chemicals are spilled on your skin, wash the contaminated area for at least 15 minutes. If it is a major spill, immediately remove contaminated clothing and wash for at least 15 minutes in a safety shower. 4. Quickly shout an immediate warning to all your neighbors in case of a fire. It is very important that everyone in the room know as quickly as possible when there is a fire. 5. All students should exit a lab in case of a fire. The lab instructor will call the Campus Police Dispatch at 222 (from any ACC phone) or 223-7999 (from an outside or mobile phone). 6. Speed is the most important aspect of helping a person who is on fire. Your nearest neighbors must respond quickly by smothering the fire with a fire blanket as soon as it appears. 76 7. Do not allow a person whose clothing or hair is on fire to move. Stop the person and quickly push them to the floor and smother the flames immediately with a fire blanket. 8. The student nearest a fire blanket should bring the blanket to a person who is on fire, and once the flames are quenched, that person should be taken immediately to the safety shower. Violations of safety rules Failure to follow the lab safety procedures may have consequences for your grade in this course. Major infractions that endanger anyone working in the lab (including yourself) may result in a grade of “F” for the laboratory portion of the course and/or being prohibited from participation in future lab courses at ACC, at the discretion of the instructor. 77 Biology Lab Safety Rules Worksheet and Information Review the safety rules listed above, and answer the following questions for your laboratory room. You can read the complete ACC science safety policy at http://www2.austincc.edu/sci_safe/ Emergencies If there is a fire, major chemical spill or other emergency call the ACC Police Dispatch as soon as possible. Tell the officer your campus and exact location in the building. Location of nearest ACC phone: __________________________________________________________ ACC POLICE DISPATCH: 222 (from an ACC phone) 223-7999 (from a mobile or other phone) If evacuation is necessary, go to the designated rally point away from this building. Directions to nearest exit: ___________________________________________________________ Location of rally point: _____________________________________________________________ Safety Equipment and Information: Information about chemicals used in this laboratory can be found in Material Safety Data Sheets (MSDSs) and in a chemical inventory located ______________________________________________________________ The emergency gas shut-off for this lab is located ___________________________________________ Shut off the gas immediately if gas nozzles or valves are damaged, or if there is a fire. Fire extinguishers are located (1) ____________________________________________________ (2) ____________________________________________________ To use a fire extinguisher, pull the pin in the handle and squeeze the handle while pointing the nozzle at the base of the flame. Fire blankets are located (1)_____________________________________________________ (2)_____________________________________________________ If you are on fire, stop, drop and roll. Let someone else get the fire blanket. A safety shower is located ______________________________________________________________ If you spill a significant quantity of chemical, especially an acid or base, on yourself, immediately stand under the shower and pull the handle. Disrobe. The instructor will evacuate the room and close the doors for your privacy. Someone of your gender will stay to help you. Stand under the shower for at least 20 minutes. You will be given clothing after the shower. 78 An eyewash is located __________________________________________________________________ If a chemical is splashed or rubbed into your eyes, you must use the eyewash for at least 20 minutes. Put your face close to the basin with your eyes held open and press the lever to start the spray. Someone will help you with this. If a person is experiencing electrical shock from touching wires or equipment, use a belt, apron, lab coat or other non-conducting material to pull them away from the electrical source. First aid kits are located (1) _____________________________________________________________ (2) ______________________________________________________ Only minor cuts and burns will be treated in the lab. Serious injuries must be treated in a medical facility. Emergency Medical Services (EMS) will be called if you are injured and are unable to take yourself to a medical facility. Dress code and personal protective equipment (PPE) While in the lab you must wear closed-toed shoes. In lab activities involving chemicals, you must wear long pants or skirts (below the knee) or a lab apron/coat (provided). You must wear goggles or safety glasses marked ANSI Z87.1 when directed to do so by the lab instructor or lab safety instructions. You must bring your protective eyewear with you to every lab class. If you forget your eyewear and the lab room does not have a pair to loan to you, you will not be able to participate in the lab and may forfeit your lab grade for that day. ACC cannot guarantee that loaned safety glasses or safety goggles are uncontaminated by microbes or chemicals. Wearing contact lenses in the lab is strongly discouraged. Students wearing contact lenses must wear safety goggles instead of safety glasses. You must tie back any long hair in labs involving open flames, and it is recommended you do so for any lab. Gloves are provided and should be worn for any lab activity. Your instructor will inform you when gloves are required rather than optional. For your safety, we recommend that you: o avoid wearing very loose clothing, especially long, loose sleeves. o wear natural fiber clothing (such as cotton or wool) because synthetic material (such as polyester) can melt onto skin in a fire. o remove watches, rings, and bracelets during lab activities involving chemicals. Waste disposal For chemical wastes, there are (i) flammable organic, (ii) inorganic, and (iii) organic waste containers located _______________________________________________________________________________ For other wastes, there are containers for o biohazards – located _____________________________________________________________ 79 o glass – located __________________________________________________________________ o other trash – located _____________________________________________________________ You must precisely follow the waste disposal procedures. Never dispose of anything in lab without prior direction from the instructor. Lab conduct DO NOT o horse around or perform unauthorized experiments o eat, drink, or chew (tobacco or gum) o bring drinks or food (even in closed containers) into the lab o pipet by mouth o taste chemicals, or directly smell chemical fumes You must follow all procedures in manuals, in handouts, and as given by the instructor. You must store backpacks, coats, and other personal items _______________________________________. We recommend that you bring as few items to lab as possible. Report broken glass and chemical spills to your instructor immediately. Lab hygiene You must clean up your individual work area/equipment and community work areas/equipment (such as sinks and balances). You must put lids back on bottles and containers immediately after use. Do not put excess chemicals back into original containers. Only dispose of chemicals and waste as directed by the instructor. Wash hands prior to leaving lab. Always assume the chemicals used in lab are corrosive or irritating. Any time chemicals come in contact with your skin, wash the affected area immediately. Labeling You must label containers/test tubes if you are using more than one container per lab. Inform your instructor immediately if a label is damaged in any way. Read all labels and pay special attention to hazard information. Disease Blood-borne diseases, such as HIV and hepatitis, can be transmitted from person to person through contact with human blood. Follow the Universal Precautions whenever exposure to human body fluids is possible. Consider all body fluids (saliva, blood, urine, feces, vomit) as potentially infected. Do not come into contact with anyone else's body fluids 80