HEP_24280_sm_suppinfo
advertisement

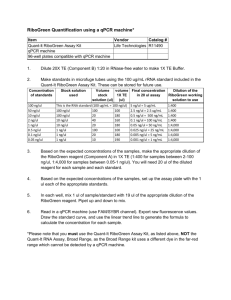
SUPPLEMENTARY FIGURE LEGENDS Supplementary Figure 1. Ectopic HNF4α expression in Hep3B hepatoma cells induces an epithelial phenotype, inhibits invasive behaviour and down-regulates mesenchymal markers (A) Phase-contrast micrographs showing the morphological changes observed in BW-H4 cells. Scale bar = 40 m. (B) Representative phase-contrast micrographs of a wound healing assay comparing BW-H4 to mock-infected BW1J at the indicated times. Scale bar = 80 m (C) Histogram of invasivity assay. Data are represented as mean and SD of triplicated experiments (P<0.01 Student t test). (D) Wound healing assay of Hep3B hepatoma cells showing after 24 hours a reduced migration of cells expressing HNF4α (3B-H4) compared to mock-infected cells. Scale bar = 80 m (E) qPCR analysis for Phosphoenolpyruvate the indicated carboxykinase, genes (HNF1, AldoB=AldolaseB, APOC3=apolipoproteinC3, PEPCK= Cdh1=E-cadherin, Krt18=Cytokeratin 18, JUP= Junction Plakoglobin, Cldn1=Claudin1, Vim=Vimentin, Fn1=Fibronectin, Mmp2=Metalloproteinase 2). Supplementary Figure 2. HNF4α induces direct transcriptional repression of human Snail gene. (A) qPCR analysis of endogenous Snail mRNA level. The values are calculated by Ct method and expressed as fold of change in gene expression in 3B-H4 versus parental Hep3B cells ( *P <0.05 Student t test) . (B) Western blot analysis of Snail, HNF4α and, as control, Tubulin in parental Hep3B and 3BH4 cells . (C) EMSA assays on HNF4α consensus binding sites of human apoC3 and Snail promoters were performed with the indicated nuclear extracts and probes. (D) qPCR-ChIP assay with anti HNF4α and anti-NCoR antibodies on chromatin from 3B-H4 cells. As controls, ChIPs were also performed with unrelated IgG. Consensus sites for 1 HNF4α, schematically depicted as black boxes (H4-2 and H4-1) and the unrelated sequences used as negative control (ns), were amplified with primers depicted as arrows and listed in supplementary TABLE 5. Mean and SD values from three independent experiments are reported with statistical significance (*P<0.05). (E) EMSA assays on HNF1α consensus binding site (-879/-862) of human Snail promoter were performed with the indicated nuclear extracts. Probes are reported in Supplementary TABLE 1. Supplementary Figure 3. Positions of HNF4α and HNF1α putative binding sites on mesenchymal genes promoters (A) Consensus sites for HNF4α on Mus musculus fibronectin promoter analyzed in qPCR ChIP assays are schematically depicted as black boxes (H4) and black arrows indicate the amplified region and the unrelated sequences used as negative control (ns). (B) Consensus sites for HNF4α and HNF1α on Mus musculus vimentin promoter and the unrelated sequences used as negative control (ns) in qPCR ChIP assays are schematically depicted as in 3A. (C) Consensus sites for HNF4α and HNF1α on Mus musculus desmin promoter and the unrelated sequences used as negative control (ns) in qPCR ChIP assays are schematically depicted as in 3A. Supplementary Figure 4. HNF4α knockout mice express mesenchymal genes qPCR analysis of the indicated genes in wild-type floxed Alb- Hnf4α (Alb- Hnf4α F/F) and AlbHnf4α KO (-/-) mice calculated by Ct method (* P<0.05, ** P<0.01 Student t test). Mean data of three different animals per group. Supplementary TABLES Legend. Supplementary TABLE 4 2 For each qPCR ChIP primers to the corresponding coding sequences were used as negative control genomic regions (ns). The primers are the same listed in Supplementary TABLE 3. Primers sequences for HNF4 target FXIIIB were previously reported 1. 3 4 5 6 7 8 9 10 11 12 SUPPLEMENTARY METHODS Cell culture conditions BW1J and Hep3B cells were grown in DMEM with 10% FBS (Cambrex BioSciences, Verviers, Belgium), 2 mM L-glutamine (Sigma-Aldrich, St. Louis, MO) and antibiotics. MMH-D3 hepatocytes were grown in RPMI 1640 with 10% FBS (GIBCO), 50 ng/ml EGF, 30 ng/ml IGF II (PeproTech Inc, Rocky Hill, NJ), 10 g/ml insulin (Roche, Mannheim, Germany) and antibiotics, on collagen I (Upstate Biotechnology, Waltham, USA) coated dishes. When indicated cells were treated with 4 ng/ml TGF (PeproTech Inc., Rocky Hill, NJ, USA) for 48 hours. Plasmids and production of recombinant retroviruses For forced expression experiments, the rat HNF4α and HNF1α cDNAs were subcloned into the pBABEpuro retroviral vector. To produce recombinant replication incompetent retroviruses, 293GP packaging cells were transiently transfected according to standard procedures with the HNF4α and HNF1α retroviral constructs together with VSV envelope protein encoding plasmid. Viral particles were collected 48 and 72 hours after transfection. Stable HNF4α over-expressing cells and control cells were obtained by infection with pBABE Flag-HNF4α orempty vector viral supernatant, respectively, and selected in 3 g/ml puromycin for a week. Site-directed mutagenesis. Mutations in each of the putative HNF4α and HNF1α binding sites on the murine Snai1 promoter were generated by site-directed mutagenesis using the QuickChange XL Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA) as indicated by the manufacturer. The point mutations in the corresponding plasmids were verified by sequencing. Primers used to generate the desired mutants are listed in Supplementary TABLE 1. Luciferase assays BW1J cells were transfected by Lipofectamine 2000 (Invitrogen, San Diego, CA) with Snail1 promoter (-900 to +21) fused to the firefly reporter gene 15 and pCLBCX-Flag-HNF4αor pCLBCX- 13 HNF1αexpression vectors. The cotransfected plasmid pCMV--galactosidase was used as control. All transfections were performed in triplicate. Luciferase activity was measured 48 hours after transfection with the Dual Luciferase Reporter Assay System (Promega Corporation, Madison, WI) and normalized for both -Galactosidase activity and protein amount. Migration and invasivity assays For the wound healing assays, a scratch was made on a confluent cell monolayer with a 200-μl pipette tip. For transwell invasivity assays, 8 m pore 24-well cell culture plates (Corning Inc, NY, USA) coated with type I collagen (0.1 mg/ml; Upstate Biotechnology, CA, USA) were used. Equal numbers of cells (25,000/well) were plated onto the top surface of the transwell filter in the upper chamber. The lower chamber contained 20 ng/ml IL-8 (Pierce, Chemical, Rockford, IL). Cells were fixed after 24 hours with 4% paraformaldehyde, stained with Giemsa solution and DAPI, and the nuclei counted manually in five random microscopic fields. Experiments were performed two times in triplicate. For the wound healing assay in HNF4α knock-down MMH-D3, cells were plated in 35 mm plates and a scratch was made 24 hours after HNF4α siRNA or siRNA control transfection. A low serum (2% FBS) culture medium was also added to the plates to inhibit cell proliferation. The micrographs were taken at 24 hours or 48 hours after the scratch on paraformaldehyde-fixed cells and the migrating cells were counted on 6 fields. Cells were also stained with DAPI to easily count the nuclei of migrating cells crossing the scratch. The assay was repeated two times. RNA extraction, Reverse Transcription and RT-qPCR Total RNA was extracted by TRIzol (Invitrogen, Carlsbad, CA) or NucleoSpin® RNA II kit (Macherey-Nagel, Germany) and reverse transcribed with MMLV-reverse-transcriptase (Promega, MI, Italy). RT-qPCR was performed using BioRad Miniopticon with KAPA SYBR® Green FAST qPCR mix (KAPABIOSYSTEMS, Woburn, MA, USA). Relative amounts were obtained with 2– ΔΔCt method and normalized to β-actin. SDS-PAGE and Western Blots 14 Cells were lysed in RIPA buffer containing protease inhibitors (Roche, Monza, IT). Protein concentrations were determined by use of the Bio-Rad assay reagent (Bio-Rad Laboratories, Hercules, CA). Equal amounts of protein were separated by SDS-PAGE and transferred to PVDF membranes (Millipore, MA, USA). Blots were blocked in 5% non-fat milk prepared in TBST and incubated overnight with the primary antibody. Antibodies against HNF4α (C19, sc6556), αTubulin (TU-02, sc8035), Snail (H-130, sc28199), Vimentin (sc-5565) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), α-SMA antibody (from Sigma-Aldrich, USA). Blots were then incubated with HRP-conjugated species-specific secondary antibodies (Bio-Rad,Hercules, CA. USA; Jackson Immunoresearch Laboratories, Inc. Baltimore, USA), followed by enhanced chemiluminescence reaction (Pierce Chemical, Rockford, IL). Electrophoretic mobility shift assays For competition experiments, nuclear extracts were preincubated with a 200-fold molar excess of wild-type or mutated cold probes. For supershift experiments, nuclear extracts were preincubated with 2µg anti-HNF4α (sc8987), anti-HNF1α (sc8986X), or anti-Tubulin (sc8035), Santa Cruz Biotechnology, as a non-specific antibody control. ChIP analysis Immunoprecipitations were performed with anti-HNF4α -NCoR (sc 8994), anti- HNF1α (sc 6547), the negative control rabbit IgG (sc-2027) Santa Cruz Biotechnology. A 1:10 dilution of starting chromatin DNA was used as template for input amplification. QPCR analysis was performed in triplicate and repeated two independent times. All signals were normalized to total input chromatin adjusted for diluition factor. Data are represented as fold enrichment of immunoprecipitates on specific target genes subtracted of background signal (immunoprecipitates with IgG) and compared to the amplification of non specific genomic regions (ns), used as negative control. For in-vivo ChIP analysis, livers were perfused with 2% w/v collagenase, dissociated in DMEM, and filtered through 70 μm strainers prior to fixation in 1% formaldehyde. Fixation was stopped 15 with the addition of excess glycine. Chromatin preparations were made using the Cell Signaling Simple ChIP Enzymatic Chromatin IP kit with magnetic beads. HNF4α was immunoprecipitated using 3 μg of goat anti-HNF4α C19 (sc-6556, Santa Cruz Biotechnology, Inc.) overnight at 4ºC. 2% input was reserved prior to immunoprecipitation. QPCR reactions were prepared using 1 μl DNA and 200 nM primers with the Fast Sybr Green mix (Applied Biosystems) and run on a 7900HT Fast Real-Time PCR machine (Applied Biosystems). qPCR ChIP primers are listed in Supplementary TABLE 4 and 5. Immunofluorescence and Immunohistochemistry. Immunofluorescence analyses were performed on deparaffinized liver sections of Hnf4 F/F and Hnf4 -/- mice. Briefly, formalin-fixed, paraffin-embedded mice liver samples were cut at 5 μM thickness and mounted on polylysine-charged glass slides. The sections were deparaffinized and rehydrated in a series of graded alcohol:water solutions and then incubated for immunohistochemistry with the primary antibodies anti-Desmin (ThermoFisher Scientific,Fremont, USA), anti α-SMA (Sigma-Aldrich, St. Louis, USA), anti-Vimentin (Millipore, Billerica, MA). Secondary antibodies were used in an indirect immunoperoxidase protocol as described previously2. For confocal microscopy antibodies against αSMA (1:200) and Albumin (Novus Biologicals, Inc., Littleton,CO; 1:100) were used. AlexaFluor-conjugated secondary antibodies were used for signal detection. Immunofluorescence analyses in MMH-D3 cells were performed with anti-Vimentin (1:50), anti-Ecadherin antibody (BD Biosciences Pharmingen, Bedford, MA; 1:50), anti ZO-1 (Zymed Laboratories, San Francisco, CA; 1:50). Supplementary References 1. Inoue Y, Peters LL, Yim SH, Inoue J, Gonzalez FJ. Role of hepatocyte nuclear factor 4alpha in control of blood coagulation factor gene expression. J Mol Med. 2006 84(4):334-44. 2. Hsu, SM, Raine, L, Fanger, H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J. Histochem. Cytochem. 1981. 29:577-580. 16 17