Lab 6 and 7 DNA sequencing
advertisement

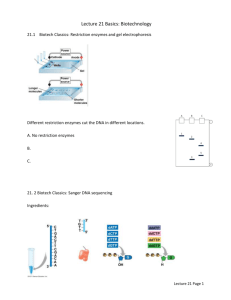
LABORATORY 6 AND 7 DNA SEQUENCING BIG DYE TERMINATOR V3.1 CYCLE SEQUENCING KIT (APPLIED BIOSYSTEMS) Text adapted by Dr. Harvey Ballard from "Nucleic acid sequencing", Chapter 9, pp. 326-339 in: Hillis et al. (1996). A. Early radioactive methods 1. Two different methods used template DNA separated into four separate reactions, isolate variously sized fragments 2. Fragments of different lengths correspond to particular positions along the template DNA 3. Fragments (100s or 1000s of copies, radioactively labeled) traveling at same rate have same nucleotide at terminated position (at least in Sanger dideoxy approach) 4. Reactions electrophoresed in four adjacent lanes, DNA samples arranged across top of solidified polyacrylamide gel 5. After vertical electrophoresis, photographic X-ray film exposed to gel 6. Sequence interpreted by reading "up" the autoradiogram (exposed, developed X-ray film) and across the four lanes, from shortest fragments (at bottom of gel) to longest (at top of gel) 7. Maxam-Gilbert (chemical) sequencing a) Cleaves DNA template at different nucleotide positions, G, A+G, T+C and C and labels these cleaved fragments b) Read A and T by interpreting double bands also including G (with A) and C (with T) c) Not used very much historically 8. Sanger dideoxy (enzymatic) sequencing a) Uses radioactively labeled primer and dideoxynucleotides (ddATPs, ddCTPs, ddGTPs, ddTTPs) b) When a dideoxynucleotide (rather than simple deoxynucleotide) is inserted into elongating DNA strand, reaction is terminated at that point c) Nucleotide sequence interpretation straightforward--A, C, G and T lanes 9. Can read up to 200 bp reliably on a very good gel (not common) B. 10. Compressions or "hard stops" common across gel where secondary structure prevents effective sequencing 11. "Ghost" bands frequent 12. Amount of time and labor per sequence, cost of isotope, failed gels or failed reactions, etc., result in hand sequencing being somewhat more expensive to do than to do automated sequencing, and less reliable! Automated fluorescent sequencing 1. Cycle-sequencing in a thermal cycler, in one tube, with a commercially prepared mixture, replaced four separate reactions using liquid nitrogen and radioactive isotope 2. Still uses ddNTP technology of Sanger method, but with ddNTPs labeled with a different colored fluorescent dye a) ddATPs labeled with a green dye b) ddCTPs labeled with a blue dye c) ddGTPs labeled with a black dye d) ddTTPs labeled with a red dye 3. Fragments electrophoresed either on a polyacrylamide gel or through a capillary (OUs machine) 4. As samples electrophorese across a slit or window, a laser "reads" the fluorescent peaks representing different-sized fragments 5. Much less time and cost in labor, fewer failed reactions, fewer problem points for "trouble-shooting" or optimization; cost of sequencing is largest expense, cost of cycle-sequencing is next one, but tiny volumes used make this relatively cheap Exercise 1. Run a gel of your products, following (modifying) the directions from Lab 1, and analyze your results. Exercise 2. Sequencing (cont.) Sequencing Step 2. Precipitation of the PCR Product for sequencing (from last lab) 1.0 µl 50 µl 20 µl NaOAc (3M; pH 5.2) 95% Ethanol PCR reaction Incubate solution for 15 min. at room temperature. Centrifuge for 20 min. at 13,000 rpm. Discard liquid, being careful not to disturb the invisible pellet. Wash with 250 µl cold 70% ethanol (4° C). Centrifuge for 5 min. at 13,000 rpm. Carefully remove the liquid. Vacuum dry for 3 min. Step 3. Submit your reactions to the sequencing facility Tour of the OU DNA Analysis Facility, 510 Porter Hall