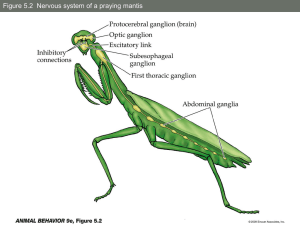
CHRONOTHERAPY OF CANCER:
advertisement

CHRONOTHERAPY OF CANCER: A MAJOR DRUG DELIVERY CHALLENGE by William J.M. Hrushesky, M.D.1 Marek Martynowicz, M.D.1 Miroslaw Markiewicz, M.D.1 Reinhard von Roemeling, M.D.2 Patricia A. Wood, M.D., Ph.D.1 The evolution of life took place in a milieu influenced by cyclic interactions of the sun, earth and moon. The existence of rhythmic changes in living organisms is a sign of their adaptation to these relationships and serves as indirect evidence for time-dependent variability of the response of the human body to many drugs, including those used in the therapy of cancer. This latter possibility has been confirmed for several classical chemotherapeutics in both murine and human trials. Doxorubicin and cisplatin, as well as their analogs, 5-fluorouracil and FUDR have been studied in the context of their circadian pharmacodynamics and toxicology. The outcomes of these studies clearly show that proper timing of their administration reduces drug toxicity and allows for substantial increases in the maximally tolerated dose, which results in better treatment efficacy and greater comfort for patients. Also, the first steps in investigation of optimal timing and scheduling of therapeutic peptides and polypeptides (erythropoietin, TNF, IL-2) have been made. Preliminary results suggest that these "natural drugs" may be considerably more circadian time-sensitive than are classical chemotherapeutic agents. The world of chronobiology provides a new dimension for drug delivery. Multi-agent therapies, where each drug will be given in a time-dependent manner, will require sophisticated computerized multiple reservoir drug delivery systems. Closed-loop, implantable devices that stipulate optimal timing according to measures of internal circadian timing are under development. Such systems will permit cancer patients to become more active and productive. Finally, the adoption of such high-tech drug delivery instruments will enable attention to be given to answering important chronobiologic questions and so will help to turn the science of chronobiology into what it truly is - a multidimensional and dynamic perspective on life and science. INTRODUCTION. Chronobiology is the study of the temporal relationships of biologic phenomena. All living things evolved in a milieu characterized by constant change based upon the cyclic relationships of the sun, earth, and moon. The early chemistry of life was strongly helio-dependent. Organisms had to store energy during periods of daylight for use during periods of darkness. Adaptability to the influence of the circadian patterns of our planet was thus a sine qua non of life and it is apparent that all organisms have incorporated and retained in their genetic make-up this essential circadian periodicity. Circadian organization is such a basic property of life that derangements may have lethal consequences, including for example, the severe effects of sleep deprivation or the major schedule disruption during occurring transmeridian travel in humans. Life forms that have evolved and remained at those parts of the earth's surface where day and night are of relatively equal duration throughout the year have developed lower frequency patterns than those that had to cope with seasonal differences in energy availability. Organisms have developed rather complex abilities to sustain themselves through long seasonal periods of energy dearth - hibernation is the example. During the millennia when life forms lived exclusively in the sea, the regular and recurrent tidal forces generated by the moon and sun acting upon the earth also required additional evolutionary adaptation of the vital chemistries of all creatures. For example, the massive and regular movements of the fluid covering the planet have defined the lunar day of 24 hours and 51 minutes, and the relationship of flood and ebb tides with spring and neap tides have defined the 29 1/2-day lunar month. Interestingly, a further rhythm having an endogenous periodicity of about 7 days (5-9 days) has been well-documented. This normally low amplitude rhythm in cytokinetic, immunologic, and other variables may be markedly amplified when the organism is perturbed. This approximately weekly rhythm is one of the most fascinating, because there is no obvious exogenous geophysical timekeeper that has set it in motion. The four biophysical rhythms - the solar day, the lunar month, the year, and the so-called circaseptan rhythm - have left an indelible imprint upon all life forms. They have created highly complex interacting temporal networks of biochemistry and genetics. To help the reader realize how strongly they affect healthy mammalian organisms, Figures 1 and 2 give some circadian patterns of such basic physiological variables as temperature and blood pressure. Chronobiology considers each of the above interacting time frames; it defines and quantifies their biological effects; and uses the understanding of such phenomenon to refine the way we ask scientific and biomedical questions as well as permits new questions to be asked. Such questions may be asked more effectively and precisely than can be done if chronobiological effects are ignored. Data will be reviewed here which show that chronobiological considerations are important for understanding cancer etiology, prevention, diagnosis and treatment. For example, in animals, carcinogenesis is dependent upon the circadian timing of carcinogen application, while disruption of the pineal-hypothalamic-pituitary-temporal balance will influence the frequency of breast cancer development. Additionally, women at high risk for the development of breast cancer have flatter circadian and circannual prolactin rhythms than do women at lower risk. Rhythmic seasonal variations in death from breast cancer and in average estrogen receptor content of human breast cancer tissue each suggest the probable importance of the low frequency rhythmic balance between host and cancer. Physiological rhythms which could serve as a basis for the time-dependent drug response of the organism. A precondition for the improvement of therapeutic index by optimal circadian drug timing is the ability to detect and quantify meaningful biologic rhythms [1], so rhythmic changes in normal organ functions have been studied extensively in murine models. A few examples of such changes follow: Cytokinetics and nucleic acid metabolism. In the mouse and rat liver, DNA synthesis, RNA synthesis, RNA translational activity, mitotic index, weight, glycogen content, and activity of numerous enzymes are all highly circadian stage-dependent and highly organized throughout the day. The circadian rhythmicities of mitotic index and DNA synthesis in rat and mouse stomach, duodenum, rectum, and bone marrow are also very well documented [2-3]. Mauer and more recently Mauer and Smaaland have shown circadian rhythms in DNA synthesis and the mitotic index from bone marrow in normal human beings [4]. Polyamines, organic anions involved in the regulation of nucleic acid synthesis [4-7], have been studied for circadian rhythmicity at the University of Minnesota's Clinical Research Center. It was found that in normal volunteers the excretion of both monoacetylputrescine and the N1/N8-acetylspermidine urinary ratio were predictably rhythmic throughout the day (Figures 3,4). These findings provide additional indirect evidence for overall circadian synchrony in the cytokinetic activity of normal human tissues [8]. Preliminary results also suggest that the circadian rhythms of polyamine excretion are disturbed in patients with cancer, indicating that either cell division patterns are disturbed or the temporal organizations of excretory organs are adversely affected. Immunological rhythms of note. The mammalian immune system is extraordinarily periodic. Circadian rhythms in all circulating blood cell types have been well documented in both experimental animals and human beings [9-10]. Numbers of total lymphocytes, B and T lymphocytes, and natural killer cells demonstrate circadian periodicity [11]. Additionally, studies of immune functions along a 24-hour scale both in vivo and in vitro have shown these endpoints to be equally circadian stage-dependent. Studies of human beings by Cove-Smith and colleagues [11] have shown that both tuberculin skin test reactivity and the incidence of human kidney rejections are circadian stage-dependent. Tavadia et al. [12] have shown that tuberculin, pokeweed-, and PHA-induced human lymphocyte transformation are circadian stage-dependent, and that the peak ability to stimulate is antiphase with the peak of serum cortisol concentration. Further, Fernandes and colleagues have demonstrated that the plaque-forming cell response of spleens from mice immunized with sheep red blood cells also has a marked circadian stage dependence [13-14]. Total RNA content of human lymphocytes has been found to have non-trivial circadian dynamics. In our laboratory, six series of blood samples were obtained from healthy volunteers and 19 series from ten women with advanced ovarian cancer. Each series included one sample at each of six equally spaced circadian stages (4 hours apart). The total RNA content per cell or per mg of cellular protein of circulating lymphocytes from normal subjects differed predictably according to the circadian stage of blood sampling. The time dependency of total RNA content of lymphocytes could best be accounted for by a 12-hour bioperiodicity. Two populations of lymphocytes (as defined by synchrony of total RNA content), or two populations of RNA, may be present in the lymphocytes of normal individuals. The first peak of total RNA content occurs about nine hours after sleep onset (time near highest circulating steroid concentration), and the other peak occurs at 18 hours after sleep onset (near to the daily cortisol low). The morphologic cell surface markers and functional activity of lymphocytes, as well as the different RNA of these subpopulations obtained at different circadian stages, need further scrutiny to clarify whether there are either two cell populations or one cell population having a bimodal RNA distribution (Figure 5). Ten women, 29-74 years of age, with metastatic ovarian tumors, and awakening daily at around 0700 hours and retiring at about 2200 hours, were admitted at monthly intervals for chemotherapy. They were studied in a manner similar to the volunteer subjects one month after treatment during the 24-hour period before the next scheduled dose of chemotherapy, on 19 separate occasions. A circadian rhythm in total RNA content of lymphocytes with a single daily peak was present in these cancer patients. The time of highest values of RNA content in the lymphocytes of these cancer patients occurred 11 hours after sleep onset (about 10:15 hours) (Figure 6) near the usual cortisol peak. Others have shown that the total RNA content of leukocytes of five healthy volunteers exhibited circadian rhythmicity [15]. The daily leukocyte RNA peak occurred at about 11:15 hours and corresponds roughly to the first daily peak in our normal control subjects. The timing of peak RNA content rhythm of leukocytes from these volunteers is very close to that of the lymphocytes of our patients. These data suggest a molecular basis for the predictable circadian differences in lymphocyte sensitivity to therapeutic manipulation. The differences in circadian lymphocyte RNA pattern between ovarian cancer patients and normal control subjects require further investigation. Metabolic rhythms of importance in drug metabolism. The reduced glutathione (GSH) content of heart muscle cells, which determine both the redox potential and salvage from free oxygen radicals, maintains a significant circadian rhythmicity [16]. This circadian organization has also been demonstrated in the nucleated cells of human bone marrow, with timing of the highest daily levels corresponding well with the daily timing of lowest doxorubicin (an important oxygen-active anticancer antibiotic) clinical toxicity. Also, important metabolic kidney functions exhibit circadian rhythmicity, and such rhythms, in part, determine renal toxicity and the excretion pattern of certain anticancer drugs [17]. Hormonal rhythms of importance in cancer disease and treatment. The activity and hormone secretion of the cells of the adrenal cortex undergo very significant rhythmic fluctuations: concentration of corticosteroids in the gland as well as the amount of these hormones in serum and 17-ketosteroids in urine show very strong and well coordinated diurnal changes. Also the contents of ACTH in rodent pituitary demonstrates a profound circadian periodicity. Cortisol concentration as well as cortisol related phenomena (i.e., blood concentration of peripheral blood eosinophils and mononuclear cells (PBM), mitotic activity of some tissues) may rhythmically modulate immunity and cell-cycle phase- specific cytotoxic (cell cycle specific) drug sensitivities of the organism. The menstrual cycle, like the circadian cycle, also has profound effects upon the balance between host and drug toxicity as well as host and development of cancer. Chronopharmacokinetics. The ability of the liver to detoxify, catabolize/metabolize a wide range of xenobiotics is circadian-stage dependent. This has been described for the liver's detoxification potential of various agents, including para-oxon, nicotine, antimycine-A, phenobarbital, hexobarbital, and cytosine-arabinoside [18-20]. Such rhythms profoundly affect the pharmacokinetics of many, if not most, useful drugs. Circadian rhythmicity in anticancer drug pharmacokinetics has been described for 5-fluorouracil, cis-diaminedichloroplatinum II (cisplatin), oxaliplatine, methotrexate, 6-mercaptopurine and doxorubicin [21-26], as well as many other agents (more detail is provided later in this text). Circadian organization of cytokinetics in tumors. Another focus of attention for chronobiology has been whether tumor cells proliferate either randomly or rhythmically. Mitotic index and/or DNA synthesis as usually measured by tritiated thymidine uptake have been used to evaluate the proliferative activity of many transplantable and some spontaneously arising tumors in laboratory rodents. The data on fast or slowly growing hepatomas illustrate the fact that tumor cell division may exhibit a more or less strong circadian organization, depending upon the stage of tumor growth in this model. Thus, well-differentiated, slow-growing tumors retain a circadian time structure, whereas poorly differentiated, fast-growing tumors tend to lose it. Such a loss of circadian rhythmicity may also be acquired during the course of tumor growth [27-28]. All in all, no consensus on their critical points has yet been achieved for either transplantable or spontaneous tumors in any species. General methodology of chrono-oncological studies. In order to interpret chronobiological data, an understanding of the methodology of chronobiologic experimentation is required. Pre-clinical chronotoxicological studies have tried to answer the question whether mice or rats tolerate the same dose of the same anti-cancer agent differently depending upon when in the day or night or throughout a 24-hour span it is given, and/or whether the LD10, LD50 and LD90 are meaningfully different when the agent is given at different times of day. These investigations are always performed in animals of the same strain, sex, and age, and which have been synchronized for at least 2 weeks in a lighting regimen usually consisting of an alternation of 12:12 hours of light:darkness in order to assure reasonable inter-individual circadian synchrony. The most widely used endpoints to evaluate the effect of dosing time upon chrono-tolerance have been survival rate, mean survival time and overall survival pattern. In other studies, organ-specific measures of lethal and sublethal toxicity have also been thoroughly investigated for most common anticancer agents. The kinds of chronobiologic study required for each agent depend upon the agent's pharmacology and pharmacodynamics. Basic chrono-oncologic study includes bolus chronotoxicology and bolus chrono-effectiveness. These types of studies determine the effect of administration time upon drug toxicity and anticancer activity when those drugs are given either by intravenous, intraperitoneal or oral bolus. For drugs which have very short half-lives, or which have more favorable therapeutic indices when given by infusion, both infusional chronobiological studies need to be performed as well as bolus studies. Such studies compare the effect of the shape of circadian-weighted infusions upon both drug toxicity and anticancer activity. Whereas bolus studies are routinely performed upon mice, infusional studies are usually performed upon rats because of size-related vascular access problems. CHEMOTHERAPEUTICS AND CHRONOTHERAPY. Doxorubicin and its analogs (preclinical data). Anthracycline antibiotics are among the most active antineoplastic agents in clinical use today. The most widely used anthracycline, doxorubicin, is a potent therapeutic agent against a wide spectrum of malignancies, but it causes substantial acute and chronic toxicity [29]. Profound myelosuppression, stomatitis, mucositis and gastrointestinal disturbances are commonly observed acute toxic effects [30]. Chronic dosing causes a cardiomyopathy at cumulative doses exceeding 500 mg/m2 [31]. In an attempt to reduce doxorubicin toxicity, new anthracycline analogues have been synthesized by slightly altering the molecular structure of doxorubicin. Among these, epirubicin (4'-epi-doxorubicin) differs only from doxorubicin in the epimerization of one hydroxyl group of the amino sugar moiety. Both the acute toxic effects and the incidence and severity of cardiotoxicity are, on a molar basis, lower for this analogue [32]. Despite their structural similarities, epirubicin and doxorubicin differ in their temporal toxicity pattern as well as in their toxicity pattern. Both molecules intercalate similarly between DNA base pairs [33], have both a similar affinity for DNA and comparable cytotoxic effects in vitro [34]. Their pharmacokinetics differ in that epirubicin is readily converted to epirubicinol, glucuronides, and aglycone compounds [35], while doxorubicin is prominently metabolized to doxorubicinol. The plasma clearance, tissue uptake and rate of catabolism of epirubicin are greater than those for doxorubicin [36], and its toxicities are proportionately lower on a weight for weight basis. The first chronotherapy studies using doxorubicin, performed in 1977, revealed that the rate of tumor shrinkage following doxorubicin treatment of a transplanted plasmacytoma in rats is dependent upon the time of day that the drug is given. Fastest shrinkage occurred when the animals were treated with doxorubicin toward the end of their daily resting span and just prior to usual awakening [37-40]. A series of six additional studies showed that the lethal toxicities of doxorubicin are circadian stage-dependent. The circadian stage of maximum doxorubicin tolerance was coincidentally shortly before normal awakening very near to the timing associated with maximal anticancer efficacy. Mormont and coworkers [41] found that administration of 25 mg/kg epirubicin as a single i.p. injection given at one of six equally spaced circadian stages resulted in 73% overall mortality from bone marrow and intestinal toxicity in mice. However, significant differences in the proportion of survivors were found, depending on the circadian stage of drug administration. Most survivors (54%) were found following injection at 06 HALO (hours after light onset) and fewest survivors (11.4%) at 18 HALO. This optimal administration time is several hours earlier than for doxorubicin, and occurs around usual mid-sleep. The same study was repeated four times during different seasons of the year, and the results were analyzed for circadian and seasonal variations in toxicity between studies. Significant effects of both circadian time and season of treatment were noted (circadian timing: F=11.9, p<0.001; season: F=24.7, p<0.001). Animals treated in late Spring and early Summer had a lower mortality rate and survived longer than those injected in the Fall or Winter (p<0.01). Best drug tolerance was calculated to be in July (Cosinor analysis; p<0.001). The circadian dependency of epirubicin toxicity was observed during all seasons regardless of the age of the animals used. Levi et al. have tested the impact of circadian timing upon toxicity for another doxorubicin analog - THP-doxorubicin, which turned out to be best tolerated in the late rest span [42-43] very near to the best time for the parent compound. Overall, based upon these data, anthracyclines should clearly be administered in the last half of the daily sleep span or just prior to usual daily awakening. Cisplatin and analogues. Cisplatin is one of the most active drugs against a large spectrum of common solid tumors. Its usefulness is limited, however, by serious toxicities including gastro-intestinal, neurotoxicity, nephrotoxicity and myelosuppression at very high doses. A variety of analogues have been developed and tested in an effort to avoid certain cisplatin dose-limiting toxicities while retaining its anti-tumor activity. Of the many cisplatin analogues developed, carboplatin has proven to be one of the most clinically useful. Its toxicities differ from cisplatin in that myelosuppression, especially thrombocytopenia, is dose-limiting while nephrotoxicity is minimal [44]. Another analogue, oxaliplatine, has proven antineoplastic activity in both experimental models and phase I/II clinical trials, lacks cross resistance to cisplatin, and demonstrates no significant hematologic or renal toxicity. Nausea and vomiting are the major dose-limiting toxicities of oxaliplatine. A recently developed analogue, B-85-0040 cells has reduced nephrotoxicity and lack of cisplatin cross resistance [45]. Its clinical toxicity and usefulness are still to be determined. Time-dependent pharmacokinetics. Underlying mechanisms for circadian changes in cisplatin toxicity include alterations in drug pharmacokinetics, with significant circadian based variations in plasma binding and urinary excretion documented for rodents as well as for humans [46-48]. However, no correlation between oxaliplatine tissue levels and toxicity has been established [49]. It has been questioned whether circadian differences in stage of cell division of target cells may play a role in the drugs' circadian toxicity profile. However, the cell-cycle dependent sensitivity of cisplatin and its analogues is poorly understood. It appears that some cells are most sensitive to cisplatin when exposed during the G1 (intermitotic) phase of the cycle, possibly because of the delay in cross-link formation, which then would be maximal during the following S phase [50]. Clinical cisplatin pharmacokinetics were studied in patients bearing ovarian or bladder cancer using an HPLC method for quantitative identification of urinary cisplatin. The pattern of urinary excretion of cisplatin was studied after 51 courses of 60 mg/m2 of this agent. Urine samples in which cisplatin was measured were obtained immediately prior to and every 30 minutes after cisplatin infusion over 4.5 hours. It was found that urinary cisplatin kinetics (peak concentration, time to peak, area under the curve) were predictably different depending upon when the drug was infused, with significantly higher concentrations, and subsequently much greater kidney damage, arising following morning administration (Figure 7) [22-23]. Murine toxicity studies. Another case in point is the pronounced circadian rhythm in cisplatin (DDP) lethal toxicity, which was demonstrated in each of a series of 11 studies over the course of about one year. Each study entailed injection of six groups of rats with toxic doses (11 mg/kg) of cisplatin at one of six equispaced circadian stages, and subsequent observation of the mortality. Each of these studies revealed that cisplatin was tolerated better when given late in the animal's active phase (Figure 8) [51]. Mortality resulted from nephrotoxicity (Figure 9) and renal damage and was most extensive in proximal convoluted tubules. A renal tubular brush border lysosomal enzyme, ?-N-acetylglucosaminidase (NAG), is released into the urine in proportion to the degree of histologically and chemically confirmed renal dysfunction induced by cisplatin. This enzyme was present in the urine in normal animals with its baseline concentration displaying a high amplitude circadian rhythm. When cisplatin was given at its least favorable time of day, the circadian rhythm of urinary NAG was maintained, but the mean and peak levels increased five-fold in direct proportion to the subsequent rise in blood urea nitrogen (BUN). When cisplatin was given at a favorable circadian time, these groups demonstrated a smaller NAG rise and had little histologic renal damage and only a small rise in BUN [52-53]. The standard method of minimizing cisplatin nephrotoxicity is to give a pre-treatment "flush" of saline. Thus, in another series of studies, an intraperitoneal saline load of 3% body weight was given to or withheld from animals concurrently with cisplatin at six separate circadian stages [54]. A marked circadian rhythm in the amount of kidney protection achieved by the fluid load was noted (Figure 10). When cisplatin or cisplatin-plus saline was given to the animals late in their activity span, a high degree of protection was found. However, when the saline flush and cisplatin were given to the animals at the circadian stages associated with early activity, less effect resulted from the kidney protection regimen. These data indicated quite clearly not only that the lethal nephrotoxicity of cisplatin was circadian-stage dependent, but also that the standard method of renal protection (hydration) was circadian-stage dependent in its ability to decrease cisplatin nephrotoxicity [53]. We have also tested whether cytotoxicity and anti-tumor activity of the cisplatin analog B-85-0040 are circadian stage dependent. We treated 167 mice with a single i.p. dose of B-85-0040 at a dose range between 300 and 525 mg/kg at one of six equally spaced circadian stages. The administration of 300 mg/kg resulted in an overall mortality of 5%. The best drug tolerance as gauged by weight loss was observed at 14 HALO (p<0.01) (Fig. 11). The administration of 525 mg/kg resulted in an overall mortality of 84% (range 53-100%; X2=11.5, p<0.05) from bone marrow aplasia and intestinal damage. The lowest mortality rate and longest survival times were observed in the groups that had received treatment between 14 and 18 HALO (F=5.4, p<0.001; Cosinor: p<0.004). Subsequently, 46 mice were treated at one of 3 different circadian timepoints (0, 8, and 16 HALO) with a single i.p. dose of 300 mg/kg B-85-0040 five days after inoculation of 1 x 106 L1210 leukemia cells. Kruskal-Wallis lifetable analysis revealed highly significant differences between the treatment groups (w = 12.2, p < 0.01). Cure rates were 67% for treatment at 8 HALO, 33% for 16 HALO, and 0% for 0 HALO (Fig. 12). Surviving animals had no evidence of leukemia when autopsied on the 58th day post-treatment. As circadian stages of maximum toxicity and maximum anti-leukemic activity differ, optimal drug timing may increase its therapeutic index of B-85-0040. Studies on drug tissue distribution patterns at 1, 24, and 120 hours after single dose B-85-0040 injection did not reveal circadian differences that would explain the above observations. The underlying mechanisms are still being investigated. Combined therapy: Doxorubicin with cisplatin and their analogues. Review of preclinical experiments. Time-dependent synergistic effects of the anti-cancer drugs doxorubicin and cisplatin have been demonstrated in tumor-bearing rats. Reduction in tumor size and in the rate of renal excretion of the tumor marker, Bence-Jones protein, varied predictably depending upon when these two drugs were given [55]. In these earlier studies, however, the drugs were tested concomitantly at one of only two circadian stages (late-rest and late-activity). It was found that animals treated with cisplatin alone or concomitantly with doxorubicin died quicker than did either untreated animals or rats treated with doxorubicin alone, indicating that the dose of cisplatin (6 mg/kg) used for this study was too high. Even so, time dependent, differential toxicity was clearly observed. Animals treated in late-rest tolerated the drug treatment far better than did those injected in late-activity. The cause of death in these studies was related primarily to the bone marrow toxicity of the anthracycline which was consistent with other observations in the mouse. Two more complete studies followed these initial investigations, using lower doses of doxorubicin and cisplatin. Drug effects upon the host and tumor were tested at 6 different circadian stages. These experiments investigated whether circadian drug timing can optimize the ability of the doxorubicin-cisplatin combination to cure cancer in a rat model. Study 1 was primarily designed to test the effect of doxorubicin as a single agent at each of 6 different circadian stages. By contrast, Study 2 was designed to test the effect of doxorubicin administered only at the best circadian time in combination with cisplatin at 1 of 6 different circadian stages, in order to find the most effective circadian-timing of this drug combination. Optimal doxorubicin/cisplatin timing tripled cure rate of this tumor. These preclinical data suggested that dosing with doxorubicin and cisplatin should be separated by about 12 hours, with doxorubicin given in the early morning (e.g., 0600) and cisplatin given 12 hours removed from this (e.g., at 1800 ) for a patient on a usual sleep-wake schedule (e.g., sleep from 2200 - 0600 ). It is critical to point out that this suggested timing of the 2 drugs is by circadian stage, not clock hour. Thus, a person on a consistently different sleeping schedule (i.e., sleep from 0800 - 1600 if a night-worker) might best receive these drugs at a different clock hour (i.e., doxorubicin at 1600 and cisplatin at 0400 for the previous example). This raises many questions about the circadian time structure of shift workers for which there are less than clear answers. All clinical studies done to date have been performed upon diurnally active and nocturnally sleeping individuals. The relative contribution of drug sequence and the span between these two agents to the schedule-dependent differences in therapeutic index was addressed in the two studies described above. The pattern of therapeutic advantage across the day was very similar in both studies, although the sequence of agents, the span between agents, and the number of courses was different in the two studies. Regardless of these schedule differences, the same doses of drug were substantially less toxic to the host, and more effective in controlling the cancer, when doxorubicin was given just prior to usual awakening and cisplatin was given in mid to late activity. We have suggested that an appropriate rhythm marker (e.g., temperature, urinary potassium) might be monitored before, during and after chemotherapy, in order to ensure synchronization and proper circadian-stage timing of the therapy [56-57]. If our results are relevant to human oncology, exploitation of circadian and other time structures for optimal cancer chronotherapeutic schedules should lead to a significant therapeutic improvement. Review of clinical data: Studies in patients with ovarian and bladder cancer. Doxorubicin and cisplatin are the most active drugs in treating several cancer types. In ovarian cancer, the combination of these drugs has an advantage over single-agent therapy when considering response rates and survival. Drug dose, to some extent, determines tumor control [58]; however, only a third of patients with advanced disease will have a complete clinical tumor response, and an additional third a partial response. In only a few cases (< 20%) will complete clinical response result in the absence of microscopic residual disease. Advanced disease is defined as ovarian cancer metastatic in the abdomen without liver involvement, FIGO* Stage III (clinical stage grouping for primary carcinoma of the ovary according to the International Federation of Gynecology and Obstetrics), or distant metastases and/or liver involvement (Stage IV). Most patients relapse and have only median survival times between 10 and 36 months [59]. These disappointing results make any possible improvement of therapy very urgent. Metastatic bladder cancer is even more difficult to treat effectively. However, chemotherapy combinations and schedules including the combination of doxorubicin and cisplatin have emerged recently that can result in complete responses in some patients. Response rates, response durations and survival patterns of the entire patient population have, however, remained unsatisfactory. Higher dosages are associated with better response rates but also with substantial toxicity; several adjuvant studies have demonstrated an increase in length of disease-free survival for chemotherapy-treated patients when compared to those who were observed following operation without treatment [60]. Clearly, our goal in chronotherapy protocols for each of these diseases was to reduce treatment-related toxicity and complication rates by optimal circadian drug timing, allowing high-doses of drug to be administered safely and most effectively. With optimal treatment timing, we also expected improved tumor control and patient survival. Toxicity study with crossover design. Treatment plan: The first clinical study was performed to test two different circadian time schedules of the same combination of doxorubicin and cisplatin with equal doses, drug sequence and interval, for possible pharmacokinetic and toxicity differences between the two agents in the same patients treated at different times of day. More than 100 monthly treatment courses consisting of doxorubicin at 60 mg/m2 and cisplatin at 60 mg/m2 were studied in 23 patients. This clinical protocol randomized initial doxorubicin treatment time between 0600 and 1800. Cisplatin followed each doxorubicin infusion by 12 hours. Each drug was infused over 30 minutes. A standard vigorous hydration protocol of 4100 mL of normal saline (20 mEq KCl per liter) preceded and followed each cisplatin infusion. Antiemetics and diuretics were not used. After the initial treatment, the timing of doxorubicin for each subsequent cycle was alternated between 0600 and 1800, so that the drug timing was crossed-over throughout the study. Twenty-one patients were considered evaluable since two patients refused further therapy after the initial treatment. Each of these 21 patients had advanced malignancy (12 had stage III and IV ovarian cancer and 9 had metastatic D2 transitional cell cancer of the bladder). To assure precision, each patient was treated in a general clinical research center metabolic ward. Cisplatin-induced nephrotoxicity: A statistically greater per course drop in creatinine clearance followed morning cisplatin administration compared to evening administration (Figure 13). This difference was most striking following the first course and then diminished as treatment time was alternated. There was either no creatinine clearance decline or a permanent 30% fall following the first dose of cisplatin depending upon when the cisplatin was given. Bone marrow toxicity: When doxorubicin was given at 0600 and cisplatin at 1800, there was less neutropenia and thrombocytopenia than when the doxorubicin was given at 1800 followed by cisplatin at 0600. The morning doxorubicin schedule resulted in statistically significantly less depressed low counts and in full recovery of all counts to pretreatment levels, usually within 21 days of treatment, while evening doxorubicin led to less than full recovery, even after 28 days following therapy. This is demonstrated in the pattern of the fall and recovery of leukocytes, neutrophils, and platelets in an individual treated four times on one circadian schedule and four times on an opposite circadian schedule (see Figures 14,15; shading represents standard errors of counts). The clinical relevance of these findings is demonstrated by the fact that treatments given with morning doxorubicin resulted in statistically significantly fewer dose reductions and fewer treatment delays and fewer serious treatment-related complications than found with the opposite circadian drug schedule. Cisplatin-induced nausea and vomiting: The most common reason for the discontinuation of cisplatin treatment is the patient's refusal to accept further therapy because of the severe nausea, vomiting and anorexia that it causes in nearly all cases. Until recently, no antiemetic regimen had proven effective in eliminating this often dose-limiting toxicity. Nausea and vomiting were studied quantitatively in 101 courses of combination doxorubicin and cisplatin chemotherapy administered without antiemetics. Those patients who received cisplatin at 0600 had more vomiting episodes (P < 0.01), which tended to begin sooner and last longer [61]. See Figure 16. Randomized non-crossover study: Cumulative drug toxicities and efficacy. Treatment plan: In the subsequent protocol, patients were randomized to receive each of the nine planned doxorubicin-cisplatin treatments starting always at 0600 (morning) or 1800 (evening). This fixed random assignment of circadian treatment stage allowed analysis of the effect of drug timing upon all acute and cumulative drug toxicities, as well as upon the effect of circadian schedule on quality of tumor response (partial and complete response rate), time to response, response duration, patient survival, and cure rate. Circadian Schedule A was morning doxorubicin followed by evening cisplatin, and Schedule B was evening doxorubicin followed by morning cisplatin. Bone marrow toxicity: Complete evaluation of the bone marrow toxicity of the first 37 patients who received all of 9 planned treatments revealed that the circadian stage of chemotherapy administration determines whether or not this combination of drugs induces cumulative bone marrow toxicity. Because of leukopenia, most patients treated on Schedule B had to have greater than 33% doxorubicin dose reduction and many of them had to have treatment delays of greater than two weeks as opposed to those on Schedule A. Assessment by linear regression analysis of individual WBC decrease and recovery (on days 1, 7, 14, and 28) after treatment revealed more cumulative bone marrow toxicity for the majority of the patients treated on circadian Schedule B than for Schedule A, despite substantial dose reductions. Cisplatin-induced nephrotoxicity: Patients bearing cancer, or with other serious illnesses, may not be precisely synchronized enough with regard to circadian rhythms that are important in determining the amount of drug toxicity the patient might experience. In order to investigate this finding more thoroughly, the circadian rhythm characteristics of body temperature, neutrophil count, lymphocyte count, heart rate, blood pressure and urinary volume, sodium, potassium and cortisol excretion were studied. Forty-three patients were studied in this way prior to 295 separate treatment courses. Creatinine clearance fall after each treatment was then compared. Less nephrotoxicity was seen when cisplatin was given at 1800, as compared to 0600. For 24 to 48 hours prior to each treatment, urine was collected every two hours and the rate of potassium excretion determined. Each individual's circadian rhythm in urinary potassium excretion (expressed as mEq per hour) was calculated for each course. The amount of subsequent renal damage was assessed by the creatinine clearance decrease prior to the next course of treatment. Mean creatinine clearance decrement results were also compared according to how far from the daily potassium peak excretion that patient had, in fact, received the cisplatin. Creatinine clearance results were analyzed according to whether cisplatin was received 0 to 6 hours or 6 to 12 hours after the daily peak in potassium excretion. This procedure compared treatment times as gauged by a measure of internal, rather than external time. Patients who were treated within three hours on either side of the span during which their rate of potassium excretion was highest suffered no subsequent loss of renal function, while those patients receiving cisplatin farthest away from the time of highest potassium excretion had an average loss of 8 mL/min. in creatinine clearance per treatment course. Since the standard treatment course of cisplatin for this group of patients included nine courses of therapy, inopportune timing of repeated cisplatin administration resulted in a substantial and preventable loss of kidney function of more than 50%. Other toxicities: neurotoxicity, chronic anemia, and transfusion requirement were each statistically significantly different in favor of morning doxorubicin and evening cisplatin [62]. Circadian schedule dependence of toxicity and dose intensity. Toxicity evaluation following each of the 247 evaluable treatment courses included weekly 8 a.m. sampling of hemoglobin, total and differential white blood cell count, platelet count and creatinine clearance. These weekly laboratory values, combined with a monthly interim history and physical examination, served to guide dose and schedule modifications. Doxorubicin dose modifications or schedule delays were forced by three types of events. These were1) a recovery (day 28) absolute granulocyte count below 1500 cells/mm3; or 2) a recovery platelet count under 100,000 cells/mm3; or 3) interim infection or bleeding If any of these conditions were present, a 25% doxorubicin dose reduction or one-week treatment delay with subsequent re-evaluation was instituted. Doxorubicin doses were more often reduced if an infection or bleeding complication supravened, and treatment delays were more common with a poor recovery of blood cell counts. No dose or schedule modifications were instituted on the basis of low counts. Cisplatin was discontinued if creatinine clearance fell below 30 mL/min., but otherwise given at full dose. Treatment complications were defined as: interim clinical infections that required oral or parenteral antibiotics; interim bleeding episodes of any kind, whether or not platelet transfusions were administered; and anemia requiring a transfusion. Each transfusion episode usually required administration of two or three units of packed red blood cells. The rates of chemotherapy-related toxicity following either treatment schedule were calculated per patient group and per treatment courses. The results are shown in Figures 17&18, clearly indicating the profound influence of the time of day of chemotherapy upon drug toxicity and maximum dose intensity. Circadian dependence of tumor response and patient survival in ovarian cancer. Sixty-three consecutively-diagnosed women (median age 60, range 29 to 87) with FIGO Stage III [48] and IV [19] epithelial ovarian cancer were treated using one of 4 temporal schedules of the same two-drug protocol (60 mg/m2 of both doxorubicin and cisplatin every 28 days for 9 months) to test whether drug timing affected tumor control. Fifteen of these 63 women had optimal debulking operations and 48 had bulky disease with residual masses (massive disease of > 10 cm masses in 40 patients). Sixty of these 63 women were evaluable for response and survival. Each of the four treatment groups was comparable with regard to patient age, FIGO stage, histological grade of cancer, and quality of debulking surgery. Of these evaluable patients, 16 women had received prior chemotherapy and 9 had prior abdominal pelvic irradiation. The four different schedules were: U: (n=11) treatment with doxorubicin and cisplatin at unspecified times of the day with no consistent sequence or interval between drugs. A: Randomization to receive doxorubicin given at 0600, followed 12 hours later by cisplatin. B: (n=20) Doxorubicin given at 1800, followed 12 hours later by cisplatin. A/B: (n=12) Patients alternated monthly on A and B. Tumor responses are shown in Table 1. Circadian scheduling significantly increased clinical complete response (CR) rates (X2 = 38.8, P < 0.001) and survival. Median time of observation of all patients exceeded 67 months (16-105 months). Pathological complete response rates were also higher with chronobiological administration. All patients treated without regard to drug timing (Schedule U) died within 3 years. Patients receiving Schedule A had a 5-year survival of 44%. This survival was exceeded by patients treated using the crossover timing regimen. Patients treated on Schedule B had an 11% 5 year survival (Figure 19). Part of this schedule-dependent difference in patient survival may have been related to differences in average dose intensity. Mean dose intensity (as % of planned dose) of each agent for Schedule B (80% for doxorubicin and 82% for cisplatin) was statistically significantly lower than for Schedule A (94% for doxorubicin and 95% for cisplatin) and for the crossover regimen A/B (89% for doxorubicin and 90% for cisplatin). However, patients on Schedule U received the drugs at the same or higher dose intensity (94% for doxorubicin and 103% for cisplatin) as patients treated with Schedule A or A/B, but their drug schedule failed very rapidly. While a difference of 10-15% in dose intensity does not appear important, it must be kept in mind that the relationship of drop in dose intensity to drop in disease control and cure rate may not be linear. It can be expected from work by Levin and Hryniuk [63] that a drop of 10% in dose intensity may result in a 50% drop in the ability of that regimen to control cancer. Therefore, this statistically significant toxicity-related drop in dose intensity may be important to our findings that timing of doxorubicin-cisplatin administration markedly affects patient survival. The observation that unspecified treatment timing results in relatively high dose intensity, but poor disease control, also needs additional comment. That is, the dose intensity formula does not take into account the possibility of early treatment failure. In reviewing the mean number of courses for patients receiving treatment at unspecified times and those receiving time-specified treatment, it is clear that early failures occurred much more frequently in the group receiving treatment at unspecified times. In fact, the mean number of treatment courses in this group is only a little more than three, whereas 8-9 treatment courses were given using all time-specified treatment regimens. The other conclusion that we must reach about dose intensity and circadian timing is that while dose intensity is important in determining how well a combination of treatment controls cancer, there may also be effects of timing that are separate from and additive to dose intensity effects. In any event, it is clear that dose intensity can be maximized by appropriate circadian treatment timing, and that optimal treatment timing results by whether mechanism(s) in prolongation of patient survival in advanced ovarian cancer. Circadian-stage dependent doxorubicin/cisplatin treatment for transitional cell bladder cancer. The same two-drug combination of doxorubicin and cisplatin was given to patients with transitional cell carcinoma of the bladder. Patients were randomized to receive the drugs at a dose of 60/m2 each at monthly courses following Schedule A or Schedule B as outlined above. Forty-three consecutively-diagnosed patients with widely metastatic cancer received up to nine monthly courses of the two-drug combination, followed by cyclophosphamide, and 5-fluorouracil together with cisplatin as maintenance for up to two years. Fifty-seven percent of the 35 evaluable patients had objective response and 23% had complete clinical responses to the treatment. Median survival from the first treatment for complete responders was more than two years, and was one year for partial responders. Three of the complete responders were alive without evidence of cancer more than two years after stopping all therapy. Our chronotherapeutic approach safely allowed application of high dose intensity treatment. The stipulation covering the order of the drugs, interval between them, and the circadian time may have been favorable factors for this treatment's success, and which compares very favorably to other chemotherapy regimens reported to date. The fact that three patients with biopsy-proven metastatic transitional cell carcinoma of the bladder have been taken off all therapy without disease recurrence may suggest the eventual chemotherapeutic curability of this disease, which was not possible before. Unfortunately, the numbers of patients per treatment group did not allow interpretation of schedule-dependent differences in drug efficacy. Toxicity evaluation confirmed, however, that Schedule A was superior to Schedule B with lower toxicity in spite of higher dose intensity. Adjuvant chronotherapy for transitional cell bladder cancer. Finally, the same two-drug combination was given to 16 patients with transitional cell carcinoma to the bladder, and who received chronotherapy monthly either on Schedule A or Schedule B immediately after radical cystectomy. In 5 patients, cancer had penetrated through the serosa of the bladder wall into the perivesical fat (stage C). In 11 patients, cancer had spread further to other pelvic organs and pelvic lymph nodes (stage D1). Eleven of these 16 patients showed no recurrence of the disease after a median follow-up time of 3.5 years (range of 1 to more than 5.5 years). Two of the 5 patients who ultimately failed the treatment becuase of its toxicity had local tumor recurrence that developed much later than is usually the case (at 37 and 42 months). The circadian-timed drug regimen, given in full doses for nine courses as adjuvant treatment, delayed and possibly prevented local and distant recurrence of the stage C and D1 bladder cancer which can otherwise be expected in more than 90% of patients within two years of the surgery [64]. Again, similar differences in schedule-dependent toxicity were observed, but the number of patients per treatment was too low to properly test for differences in antitumor activity. Circadian timing of THP-doxorubicin with cisplatin. Levi and coworkers [65] reported the efficacy and toxicity of the new anthracycline, 4'-0-tetrahydropyranyl doxorubicin (THP) (50 mg/m2 intravenous bolus) in association with cisplatin (100 mg/m2 as an intravenous 4-hour infusion) in 31 patients with advanced ovarian carcinoma. Twenty-eight patients were assessable for toxicity, 25 for response. Nine patients had received prior treatment. Patients were randomized to receive schedule A (THP at 6 hours, then cisplatin from 16 to 20 hours) or schedule B (THP at 18 hours, followed by cisplatin from 4 to 8 hours). Schedule A was hypothesized as less toxic since THP was best tolerated in the late rest span and cisplatin near the middle of the activity span in experimental studies [42-43]. The rate of clinical complete responses was 52%, that of partial response was 12%. The overall clinical response rate was 64% (schedule A 73% and schedule B 57%). The progression-free survival and overall survival times were 10 and 19 months, respectively. Schedule A was associated with less neutropenia (p=0.1), thrombocytopenia (p<0.01), anemia (p<0.01), and renal toxicity (p<0.05) than schedule B. Of four patients withdrawn for toxicity, three were on schedule B (one death). Mean dose intensities of THP and cisplatin decreased by 30% and 47% over the five initial courses, respectively. Such decrease was significantly more pronounced for schedule B than for schedule A in previously untreated patients (p<0.01). The authors concluded that THP-cisplatin toxicities can be significantly decreased by dosing THP in the early morning and cisplatin in the late afternoon as compared to the opposite times of drug administration for both drugs. These findings confirm our earlier results, described above. Mechanisms of circadian drug pharmacodynamics of doxorubicin and cisplatin. Doxorubicin-related heart muscle and bone marrow toxicities and pharmacodynamics are believed to be a result of its NADPH-dependent single electron reduction to a reactive metabolite. In both mice and man, the availability of a key free radical scavenger, reduced glutathione (GSH), is circadian-stage dependent, with peak levels at the time of lowest drug toxicity [16,66]. Tumor response, however, occurs mostly secondary to drug binding to DNA intercalation and single strand breaks. Both circadian changes in cell cytokinetics (e.g., rate of cells in S-phase DNA) and tissue-specific differences between tumor and normal organs may have determined the observed circadian differences in doxorubicin therapeutic index. Furthermore, circadian changes in drug pharmacokinetics may have influenced the toxicity rhythm. Both the normal kidney function and cisplatin pharmacokinetics are circadian-stage dependent [67,68]. Pronounced circadian rhythmicity in lethal toxicity due to cisplatin was demonstrated in each of a series of eleven studies over a course of about one year. Each study entailed injection of six groups of rats with a toxic dose (11 mg/kg) of cisplatin at different circadian stages and subsequent observation of the mortality. It was found that cisplatin was tolerated far better when given late in the animals' active phase [52]. It was also shown that mortality resulted from nephrotoxicity. This was proven by monitoring blood urea nitrogen (BUN) and by microscopic section of the severely damaged kidneys. Kidney damage was most extensive in the proximal convoluted tubules. A brush border lysosomal enzyme, NAG, was found to be released into the urine proportionately to the degree of renal dysfunction induced by cisplatin. This enzyme was present in urine in normal animals, and its baseline concentration was found to display a high amplitude circadian rhythm. When cisplatin was given at the time it was most toxic, the circadian rhythm in urinary NAG was maintained, but the mean peak levels increased 5-fold in direct proportion to the subsequent rise in BUN. If cisplatin was given at a favorable circadian stage, these rats did not demonstrate much NAG rise and had little renal damage with only a small rise in BUN. Mortality from doxorubicin, daunomycin, epirubicin and cisplatin is also influenced by a non-trivial seasonality. For cisplatin, the extent of predictable seasonal variation in the mean survival duration of rats after drug injection was about 40 percent [69]. These findings reflect the importance of circannual rhythms at all levels of biologic organization. It is especially interesting that these circannual susceptibility rhythms are found in laboratory animals kept under constant conditions [70]. It is not unreasonable to expect that they may be greatly amplified in free-living animals. Fluoropyrimidines. Trials of mechanistic importance. DNA synthesis is interrupted because of the biding of FdUMP to thymidylate synthase (TS). This binding is tightened by intracellular folate which complexes with the FdUMP/TS. RNA production is affected because of the incorporation of the fluoropyrimidine triphosphate into abnormal RNA. Fluoropyrimidines anti-metabolites interfere with DNA, RNA, and protein synthesis. They are widely used cytotoxic drugs. The toxicity pattern is dependent upon dose and administration mode (e.g., bolus injection or continuous infusion). Myelosuppression, stomatitis, and intestinal toxicity are dose limiting, and we have found that circadian timing of these agents markedly affects their toxic/therapeutic ratios. Animal trials. DPD-activity in liver cells. The activity of dihydropyrimidine dehydrogenase (DPD), the initial, rate-limiting enzyme in fluoropyrimidine catabolism to non cytotoxic products, was measured by Harris et al. [71] at various times over a 24-hr period in the livers of male Sprague-Dawley rats housed under standardized conditions of light and dark. Under "normal" conditions, i.e. lights on from 6:00 AM to 6:00 PM and off from 6:00 PM to 6:00 AM, a circadian rhythm of DPD activity was observed (p<0.0001, cosinor analysis) with the peak of activity at 4:00 PM or 10 HALO (2.96 nmol catabolites/min/mg) and the trough at 4:00 AM (0.40 nmol catabolites/min/mg). Maximum enzyme activity exceeded minimum activity more than 7-fold. Reversing the light-dark cycle resulted in a corresponding shift in enzyme activity. Catabolism of FU in the isolated perfused liver of the rat. The same group subsequently measured the catabolism of 5-fluorouracil (FU) in an isolated perfused rat liver model (IPRL) at various times of the day [72]. IPRLs were prepared from rats sacrificed at 3-hour intervals and the elimination rate of FU and FU-catabolites (i.e., rate leaving the IPRL in the effluent perfusate) following infusion of FU was analyzed for circadian periodicity. Animals were housed under standardized conditions of 12 hours of light and 12 hours of dark. A significant circadian rhythm was observed in the elimination rate of FU and FU-catabolites, with 40-60% variation around the mean (p<0.001, cosinor analysis). Under "normal" light conditions, the peak and trough elimination rates of FU were at 19 HALO and 7 HALO, respectively. There was a reciprocal relationship between the elimination rates of FU and FU-catabolites (Table 2). Under the reverse light-schedule conditions, the rhythms were also reversed. Such a variation in the hepatic elimination rate of FU, if also present in humans, could result in a variation in the systemic level of drug during chemotherapy, thus affecting the therapeutic efficacy of FU. Time dependent drug interconversion. We adapted 60 female F344 Fischer rats to a 12:12 hour light:dark schedule. They were kept in fully climatized, sound-proofed vaults with food and water freely available. Continuous infusion was achieved by cannulation of the tail vein with a catheter, by the method described by Danhauser et al. [73] They were randomized to receive 350 mg/kg FUDR as a 6-hour constant rate i.v. infusion by Intelliject pump from 10-16 or 22-04 HALO, the most or least toxic circadian stages found in our previous rat toxicity studies. Blood was collected at 0, 2, 4, and 6 hours of infusion and at 5, 10, 15, and 30 minutes thereafter (4 samples per timepoint). FUDR plasma levels, as well as FU plasma levels (as FUDR is converted into FU), were determined by gas-chromatography/mass-spectrometry (GCMS) with a sensitivity limit of 1ng/ml (Table 3). The maximum plasma concentration (Cmax) of FUDR occurred at the end of infusion; Cmax of FU was 10-15 min. delayed and was more than twice as high at 10 to 16 HALO as compared to 22 to 04 HALO infusion (t=1.3, p=0.2). The proportion of FUDR converted into FU was greater after infusion given between 10 and 16 HALO (48 vs 25%; t=1.4, p=0.2). These findings suggests that the activity of the converting enzyme DPD is circadian stage dependent. The half-life (t1/2) of FUDR was prolonged at 16 HALO (t=1.04, p=0.3) (Figure 20). This pilot study suggested alterations in FUDR catabolism at two different circadian stages. Possibly due to small sample size, they did not reach statistical significance. Because only two circadian stages were tested in this study, the maximum circadian variation may occur at different stages and may be far greater. Human trials. Circadian changes in DPD activity in human mononuclear cells. Tuchman et al. [74] studied 3 healthy male and 4 female, 24 to 41 year old volunteers. All 7 individuals had typical sleep and activity patterns and shared a similar diet during testing. Peripheral blood was drawn every 3 hours (5 cases) or every 6 hours (2 cases) for 24 hours. Enzymatic DPD activity in cytosolic preparations of mononuclear cells was assayed with high performance liquid chromatography (HPLC) and radioactivity flow monitoring. The specific activity of DPD was clearly circadian-stage dependent. Each individual showed as little as a 38% and as great as a 99% range of values throughout the 24-hour observation span. The peak enzyme activity was consistently located around midnight for each subject and rhythmometric analysis described a statistically-significant group rhythm with predicted time of peak values occurring between 10 PM and 2 AM. The variation around mean ranged from 20 to 60.8%. These findings were independently confirmed by Harris et al. [75], who simultaneously determined the activity of DPD in peripheral blood mononuclear cells and plasma concentration of FU in cancer patients receiving FU by protracted continuous infusion (300 mg/m2/day over several weeks). Blood samples were drawn every 3 hours over a 24-hour period, after the patients had received continuous FU infusion for more than 2 weeks. The resulting DPD and FU values were analyzed for circadian periodicity. In the 7 patients studied, a circadian rhythm of DPD activity was observed (p < 0.00001, cosinor analysis) with the peak of activity at 1 a.m. (0.20±0.01 nmoles/min/mg) and the trough at 1 p.m. (0.11±0.01 nmoles/min/mg). In addition, a circadian rhythm was observed for the plasma concentrations of FU obtained over a 24-hr period (p < 0.00001, cosinor analysis) with peak values (27.4±1.3 ng/ml) occurring at 11 a.m. and trough values (5.6±1.3 ng/ml) occurring at 11 p.m. The ratio of the maximum concentration of FU to the minimum concentration observed was almost 5-fold. These studies demonstrated a circadian variation of DPD activity in human peripheral blood mononuclear cells and a circadian variation of FU plasma levels in patients receiving the drug by protracted continuous infusion. They also revealed an inverse relationship between DPD activity in peripheral blood mononuclear cells and plasma FU concentration. Circadian variation of FU plasma level in patients receiving combination chemotherapy. Petit et al. [76] demonstrated a circadian rhythm in the plasma concentration of FU in seven patients with bladder cancer receiving this drug as a continuous venous infusion at a constant rate for 5 days in combination with cisplatin. Cisplatin (45-91 mg/m2) was administered on day 1 as a 30-minute i.v. infusion at 5 PM. The FU infusion (450-966 mg/m2/day) was started on day 2 at 8:30 AM via a volumetric pump, and lasted for 5 days (until day 6). Blood samples were obtained every 3 hours on days 2, 4 and 6 for each patient (20 samples/patient). FU plasma concentration was determined by HPLC. Mean lowest and highest values (±SE) were, respectively, 254±33 ng/ml at 1 p.m. and 584±160 ng/ml at 1 a.m. (F=2.3; p<0.03). Because of large inter-subject differences in 24-hour mean plasma concentration, data were also expressed as percentages of each patient's 24-hour mean. Both analysis of variance and cosinor analysis further validated (p<0.0001) a circadian rhythm with a double amplitude (total extent of variation) of 50% of the 24-hour mean and an acrophase located at 1 a.m. (estimated time of peak). This peak occurred at a different time as compared to Harris' [75] patients. However, drug schedules and doses differed greatly in the two trials. The cisplatin chemotherapy, which preceded the FU infusion, may have affected the rhythmic cycle of FU plasma levels in Petit's study [76]. Table 4 summarizes the clinical data on circadian changes in DPD activity and corresponding FU plasma levels. Review of preclinical chronotoxicological experiments. 5-Fluorouracil. Lethal toxicity. Although fluoropyrimidines are now commonly used by long-term infusion and, although the toxicity pattern of bolus and infusional administration are disparate, most screening toxicology has been done using bolus administration. Several investigators have tested whether FU toxicity is circadian stage dependent. Burns and Beland [77] used a dose escalation protocol in male CD-1 mice standardized to a 12:12 hour lights on:off schedule. The animals were randomized to receive the treatment at one of two circadian stages: 5 HALO (mid-sleep); or 17 HALO (mid-activity). They found drug tolerance to be significantly different at these two times. The single intra-peritoneal (i.p.) bolus dose of FU killing 50% of the animals (LD50) was significantly higher at 5 HALO (450-500 mg/kg) than at 17 HALO (250-300 mg/kg). Nearly a 2-fold difference in lethal toxicity, depending upon which time of day the FU was given, was found. Similar observations were made by Popovic et al. [78] who studied the tolerance of female C3H mice for single i.p. FU bolus injections of 200 mg/kg at one of six different circadian stages (lighting schedule not given). The best drug tolerance with 30% mortality was at "mid-rest phase of the animals," corresponding to 5-7 HALO. One-hundred percent mortality was observed for treatment during the late activity phase of these nocturnally active animals' circadian cycle (approximately 20-22 HALO). These two studies each showed that the best timing for FU was in the rest phase several hours prior to usual awakening. Gonzalez et al. [79] tested lethal toxicity of single i.p. FU bolus injections and, concurrently in a second experiment under exactly the same conditions, that of 5-fluoro-2'-deoxyuridine (FUDR) i.p. bolus injections at one of six different timepoints in Balb-C mice, standardized to a 12:12 hour lights on:off schedule. Lethal toxicity at an FU dose of 150 mg/kg i.p. was significantly different dependent upon its circadian treatment time. Maximum lethality was 100 % at 2 HALO; minimum mortality was 13 % at 10 HALO (analysis of variance (ANOVA), F=11.9, p<0.001; Kruskal-Wallis life-table analysis, w=38.9, p<0.001}. These 6-timepoint studies demonstrated that the least toxic time of day for FU was two hours prior to usual daily awakening. These 3 studies performed at separate locations are quite consistent in their findings with regard to the optimal time of bolus FU to lower serious toxicity. FU should be far safer if given by bolus between mid-sleep and 2 hours prior to usual awakening. Bone marrow toxicity and anticancer activity. The time-dependencies of specific non-lethal fluoropyrimidine toxicities have also been investigated. Peters et al. [80] tested the related anti-tumor and anti-bone marrow activity of FU in transplantable mouse-colon carcinomas Colon-26 in Balb-C mice and Colon-38 in C57Bl/6 mice, standardized to a 14:10 hours lights on:off schedule. The animals were randomized to receive multiple i.p. doses at either early rest or late rest phases of their circadian cycle (2 or 12 HALO) (Balb-C: 100 mg/kg/day on days 0,7, and 14; C57Bl/6: 60 mg/kg/day on days 0 and 7 for toxicity evaluation and on days 0,7,14, and 21 for anti-tumor activity testing). Unfortunately each of these two test times missed what would have been predicted to be the most and least toxic times of day according to the work of the above 3 predecessors. Increased anti-tumor activity was none the less reproducibly found at 2 HALO compared to 12 HALO, especially against Colon-38. Hematotoxicity was also evaluated in C57Bl/6 mice for the same circadian stages. Leukopenia was only observed after treatment at 12 HALO but not at 2 HALO. Therefore, the therapeutic efficacy of bolus FU was markedly improved by treating at 2 HALO. This is consistent with a safer and more effective treatment timing prior to usual daily awakening. Intestinal toxicity. Gardner and Plumb [81] used Wistar rats standardized to a 12:12 hour lights on:off schedule for toxicity studies at multiple circadian stages. Food and water were freely available. The impairment of water absorption by small intestine in vitro, and the incidence of diarrhea in vivo were assessed after the same single i.p. dose of FU (187 mg/kg) as a function of circadian treatment time. Their findings were correlated to deoxyribonucleic acid (DNA) content of small bowel epithelium homogenate at the same circadian stage of treatment. Impairment of water absorption, the incidence of diarrhea, and weight loss, were minimal after treatment at mid-daily activity (19 HALO). The DNA content of mucosa of the small intestine also showed marked diurnal variation, with a maximum at 20 HALO. During this stage, the maximum number of cells are in the post-mitotic resting (G1) phase, and are less susceptible to the effects from the antimetabolite FU. It is noteworthy that diurnal variations in various intestinal absorptive and enzymatic activities and in the proliferative state of the epithelium have been shown to be related to the feeding time in rats; the lowest DNA synthesis rate appears to occur at or after feeding. Table 5 gives an overview of FU circadian pharmacodynamics. The mid-activity optimum for the intestine to tolerate bolus FU is several hours later in the day than is the optimum time for whole animal toxicity and lethality. This observation requires further study in both fasted and fed animals and in single and multiple dose regimens. It raises, however, the possibility that optimal times may be different for different target tissues as well as for different modes of fluoropyrimidine delivery. Recent FU chronotoxicological study. In our recent trial we have studied the time-dependent gastrointestinal, bone marrow and overall toxicity of 5FU in 150 CD2F1 female mice. We have administered 200 mg/kg FU i.v. as a bolus injection at one of six equispaced circadian timepoints to each animal. We have found that toxic effects were significantly dependent upon circadian treatment time. Maximal mortality (70%) took place in mid-activity (considered as a time of maximal DNA synthesis [77]), while 100% of animals treated in an early rest have survived the FU treatment (p<0.001). These data correspond to the evaluation of toxicity by body weight loss and recovery following FU administration, calculated as percentage of initial body weight for each animal. The largest loss of body weight was observed during mid-activity, while the most stable weights and the best recoveries were observed at the mid-rest (considered as time of minimal DNA synthesis) [77] and at an early rest time of a day (p<0.001) respectively. Circadian dependency of gastrointestinal toxicity was evaluated on the basis of weight of feces collected within 24-hour periods. Lowest values, suggesting lower food consumption and impairment of the daily production of stool, occurred when animals were treated during their late and mid-activity phases, while treatment earlier in the daily activity span resulted in the lowest decrease in weight of stool ( p<0.001). Analysis of data regarding bone marrow toxicity was based on an estimation of white blood cells levels. This showed significant circadian timing-dependent differences. White blood cells remained almost untouched after treatment in mid-rest and at mid-activity spans. Recovery at these two timepoints was also excellent. The deepest depression of white blood cell count was observed when the drug was given in the late-activity span (22 HALO; p<0.001). FUDR. FUDR lethal toxicity. In a series of murine experiments we explored whether FUDR toxicity and efficacy are circadian stage-dependent. Single bolus FUDR injections of 1 of several doses between 1000 and 2000 mg/kg were given at 1 of 6 equally-spaced circadian stages to more than 300 CD2F1 mice standardized to a 12:12 hours light/dark schedule. Food and water were provided ad libitum. Temperature and humidity were held constant in sound-proofed vaults. Animals were followed for survival every 4 hours. Survival varied predictably by more than 50%, depending upon the circadian stage of injection. Treatment during mid-to-late activity span (18-20 hours after light onset = HALO) was consistently associated with best drug tolerance in three studies. To identify the cause of death in these animals, we performed autopsies, macroscopic inspections, and routine light microscopy of slides prepared from liver, small bowel, colon, and bone marrow. Pathological changes were found in both small and large bowel, explaining diarrhea and dehydration prior to death. Diffuse necrosis in the liver and bone marrow aplasia were also present. Gonzalez et al. [79] confirmed that lethal toxicity of 2000 mg/kg i.p. FUDR by single bolus injection in female Balb-C mice is significantly different dependent upon the circadian treatment time. In that study, minimum mortality was 13 % at 2 HALO (ANOVA, F=2.69, p<0.03; Kruskal-Wallis life-table analysis, w=15, P<0.01); this is approximately 6 hours later than the optimal time observed in the studies described above in CD2F1 mice. However, it was also low with 20% mortality at 18 HALO, the least toxic stage in our previous studies. Table 6 summarizes all the similarities and difference in the results of all toxicity studies using FUDR by single bolus injection. Figure 21 shows percent of mortality in six groups of animals treated in different times of the day. Overall, drug tolerance was consistently better when FUDR was given during the late activity/early rest phase of the recipient. It must be concluded that the optimal circadian timing for bolus FU and bolus FUDR is not identical. It is safest to give FU by bolus some time after mid-sleep and prior to usual awakening, while it is safest to give bolus FUDR late in the daily activity span prior to sleep onset. While the two drugs are related to one another and inter-convertible it should not be unexpected that their optimal administration times differ substantially. Different metabolic pathways are responsible for activation of FU and FUDR, and the activity of regulating enzymes may follow different temporal patterns. Fluoropyrimidines are frequently given by continuous infusion. In order to test if circadian drug timing is also important for toxicity and anti-tumor activity when 5-fluoro-2'-deoxyuridine (FUDR) is given by continuous infusion, we compared seven equal dose FUDR daily infusion patterns in female Fisher 344 rats [82,83]. For the variable rate infusion, the daily dose was divided into four 6-hour portions of 68, 15, 2, and 15% to achieve a quasi-sinusoidal pattern. Drug was delivered either by constant rate infusion or by variable rate infusion with peak drug delivery at one of six different times of day. At a dose level resulting in 50% overall mortality, lethal toxicity differed substantially depending upon the circadian stage of maximum drug delivery. Variable rate infusions were more, less, or equally toxic compared to constant rate infusions, depending upon the circadian stage of maximum drug flow. FUDR lethality was lowest when variable rate infusion peaked from 22-04 HALO during the late activity/early rest span of the recipients (Figure 22). This is largely consistent with earlier bolus studies, but the best time for drug tolerance was slightly later. The circadian pattern of variable rate infusion also determined the antitumor activity in adenocarcinoma bearing rats. At a therapeutic dose level, and at identical dose intensity, the variable rate infusion pattern with peak drug flow during the late activity/early rest span resulted in significantly greater tumor growth delay than observed with either the flat infusion or other variable rate patterns. In fact, this particular pattern was the only one associated with significant objective tumor shrinkage. Therefore, FUDR toxicity and anti-tumor activity each depend upon the circadian timing of the infusion peak when given by variable rate. Since some of the studied circadian-stage related infusions are toxicologically and therapeutically inferior to constant rate infusion, the circadian pattern, and not the quasi-intermittency of circadian FUDR administration, is primarily responsible for these pharmacodynamic differences. The optimal input profile reduce FUDR toxicity and to maximize anti-cancer activity is daily infusion given late in the daily activity span. These optimal anti-cancer peak FUDR infusion times are, however, not the same as optimal FU bolus times for cancer control found for the mouse model partially investigated by Peters [80]. The best anti-cancer activity for FU in the mouse colon cancer was also close to its least toxic time of day described before (between mid-sleep and awakening). To summarize, the best time for FUDR by bolus or 48-hour infusion in terms of low toxicity and high anti-tumor efficacy is late in the daily activity and in the early rest span. The best time for FU by bolus is between mid-sleep and awakening. No infusional preclinical data currently exist in this respect. Clinical chronotoxicological experiments. 5-Fluorouracil. Limited clinical experience is available with circadian stage specified FU administration. Levi and coworkers [84] have extrapolated the quoted preclinical findings to an assumption that FU is better tolerated during the mid-to late rest phase of the recipients. There are no randomized clinical trial results yet to support of this hypothesis, however, the following preliminary Phase I data are interesting. High-dose intensity, circadian stage modulated 5-day FU infusion. Levi et al. [84] have treated 35 ambulatory patients with metastatic colorectal cancer, with 334 courses of FU by continuous intravenous infusion. A chronopump (Autosyringe, USA) or a Intelliject pump (Ivion, Englewood, Colorado, American suppliers; Aguettant, France, European suppliers) were used to automatically increase and decrease the infusion rate of FU from 0% to 200% of the 24 hour mean. Maximum drug infusion occurred at 4 a.m. and minimum at 4 p.m. The infusion was given for 5 days every 3 weeks. Intra-patient dose escalation by 1 g/m2/course was planned from the starting dose of 4 g/m2/course. Dose-limiting toxicities (WHO grade II or higher) included stomatitis (in 9% of the treatment courses), diarrhea (5%), neutropenia (4%), hand-foot syndrome (3%), and anemia (2%). No cardiac toxicity was encountered. Median maximal tolerated dose per course was 8 g/m2 in patients with good performance status, and 6.5 g/m2 in patients with impaired performance status (bed-ridden for 50% or more during the day (p < 0.01)]. Ten partial responses (30%) and 15 stabilizations [44%) occurred in 34 patients. In 22 previously untreated patients, 8 partial responses (36%) and 9 stabilizations (41%) were observed. Median survival exceeded 20 months. The authors concluded that compared to historical controls using 5-day constant-rate infusion, automatic circadian chronotherapy with FU permitted an increase of dose intensity by ~75% with fewer side effects. This may improve survival of patients with metastatic colorectal cancer. Randomized time-specified comparisons of maximum tolerated dose are clearly indicated. Circadian patterned 5-day infusion of Fluorouracil+Leucovorin+Oxaliplatine. Levi [85] has recently reported the highest objective response rate (66%) achieved to date in patients with metastatic colorectal cancer treated with intravenous, time-specified delivery of FU, tetrahydrofolate (Leucovorin = LV), and Oxaliplatin (L-OHP). In a 5-day chronotherapy protocol, the infusion rates of FU, LV and I-OHP were automatically increased and decreased along the 24 hour scale using a multi-channel pump (Intelliject, Ivion Corp., Englewood, CO). Daily doses were: FU = 500 mg/m2, LV = 300 mg/m2, and I-OHP 25 mg/m2 (3,500, 1,500 and 125 mg/m2 per course, respectively). An admixture of FU and LV was infused between 10 p.m. and 10 a.m. The infusion pattern had a bell-shape with peak at 4 a.m. Oxaliplatin was infused from 10 a.m. to 10 p.m. in a similar pattern, peaking at 4 p.m. Of 36 patients with metastatic colorectal cancer accrued to this trial since January 1988, 32 are evaluable for response (1 toxic death, 3 deaths before 2nd course in patients with PS 3 III). Eighteen patients had received prior chemo and/or radiotherapy. In 120 courses, dose-limiting toxicities included moderate vomiting (3 2 episodes/day despite 100 mg/day alizapride) and diarrhea (3 5 stools/day for > 7 days despite 10 mg/day loperamide). Reversible peripheral neuropathy with moderate functional impairment was observed in 3 patients after 3 8 courses. Disease progressed in 6 patients (19%), was stabilized in 5 (16%), and regressed objectively by >50% (assessed using computer-assisted tomography scans) in 21 (66%). If analyzed by pretreated and untreated patients, the objective response rates were 57 and 72%, respectively. Median survival has exceeded 18 months in previously untreated patients. Time-specified randomized studies of this 3-agent combination are clearly indicated in order to reproduce the results and determine what portion of the benefit of this therapy derives from optimal circadian timing and what portion derives from the combination of the agents irrespective of timing. FUDR. Randomized Phase I evaluation of circadian FUDR infusion shapes. Continuous long-term FUDR infusion frequently causes severe and dose limiting gastrointestinal toxicity when given by constant rate at commonly prescribed dose levels. Based upon preclinical data reviewed above, we assumed that FUDR would be better tolerated when most of the daily continuous infusion is given in the evening (during the late activity phase). Therefore, we compared a circadian patterned variable rate infusion with a maximal drug flow rate in the late afternoon/early evening and minimum flow rate during the early morning hours with a constant rate infusion in 54 patients with widespread cancer [86]. All FUDR infusions were administered using a programmable implanted drug pump (Synchromed, Medtronic Inc., Minneapolis, MN). In a pilot cross-over study and a second randomized trial, patients with metastatic malignancies treated with equal dose intensities experienced less frequent and less severe diarrhea, nausea and vomiting following variable rate infusion. In a third study, the dose intensity of variable rate infusion was stepwise escalated to determine the maximum tolerated dose. Patients receiving time modified FUDR infusion tolerated an average of 1.45 fold more drug per unit time while experiencing minimal toxicity. FUDR infusion was found to have activity against progressive metastatic renal cell cancer: 23% of the patients responded objectively. These studies suggested that increased dose intensity achieved by optimal circadian shaping may improve the therapeutic index of infusional FUDR and may help control malignancies such as renal cell cancer that are refractory to conventional chemotherapy [87]. Hepatic arterial FUDR infusion. Liver metastases from colorectal cancer can be treated effectively with prolonged intra-arterial infusion of FUDR. This treatment is, however, frequently accompanied by severe and sometimes irreversible liver toxicity. Fifty ambulatory patients were treated with intra-arterial infusion of FUDR at 0.1-0.3 mg/kg/day x 14 days every four weeks by implanted pump [88]. Twenty-four patients received constant rate infusion, and 26 received circadian modified infusion. Circadian-modified infusion was technically as easily achieved as constant rate infusion by using programmable pumps. Toxicity in the form of cholestasis and subsequent jaundice was less frequent, less severe, and occurred later, when circadian infusion was employed. Cholestasis gauged by alkaline phosphatase elevations occurred in 65% versus 44% of the patients and jaundice in 26% versus 4% of those receiving flat or timed infusion, respectively. Patients receiving the circadian-modified infusion tolerated 70% higher average dose intensity. Nausea, vomiting, and diarrhea were mild and occurred in 20% of the patients of each group. Gastric ulcers were equally frequent (12%). More than half of the patients (52%) had no toxicity with circadian infusion, while only one-fifth (22%) of those receiving constant rate infusions were free of toxicity (p<0.03). The objective response rates were comparable at 35% and 32% for flat and circadian infusion, respectively. Our study demonstrated that the maximum tolerated dose intensity of intrahepatic arterial FUDR infusion can be increased substantially by circadian modification of the continuous infusion rate, and debilitating liver toxicity with jaundice can be significantly diminished. Incidentally, another group of 7 patients with progressive metastatic renal cell cancer confined to the liver, who were treated with the same hepatic arterial FUDR infusion schedule, had one complete and three partial responses (objective response rate of 57%). This indicated excellent anti-cancer activity of our treatment schedule in this highly chemotherapy-refractory malignancy. Mechanisms of circadian variations in fluoropyrimidine pharmacodynamics. While much is known about fluoropyrimidine metabolism, little information is available on circadian alterations in these complex biochemical processes. The following description outlines the major metabolic pathways as a background for the circadian studies presented below (Figure 23). FUDR activation. After parenteral administration, FUDR utilizes the pyrimidine nucleotide transport system to gain entry to cells [89]. FUDR is converted in a single step reaction into the active metabolite FdUMP by deoxyuridine kinase (DUK), an enzyme with limited capacity [90]. It is not known whether drug uptake, retention, or DUK activation vary during the day. The intracellular nucleotide fluoro-deoxyuridine-mono-phosphate (FdUMP) has a relatively prolonged half-life and persists in a profile characteristic of individual tissues [91]. FdUMP competitively blocks the activity of thymidylate synthetase (TS), which converts deoxy-uridine-mono-phosphate (dUMP) into deoxy- thymidine-mono-phosphate (dTMP), thereby interfering with DNA synthesis. It is not known whether TS activity is circadian stage dependent in most tissues relevant to FUDR toxicity or in tumor. Cytotoxicity is a direct function of the amount of FdUMP and the length of time during which it is present in the cell. The blockage of TS can be reversed by increased dUMP levels, decreased FdUMP levels, decreased binding affinity to the enzyme, or increased enzyme activity. Blockage of the enzyme is greater in the presence of tetrahydrofolate excess. It is currently unknown whether tetrahydrofolate levels vary with time-of-day. The complex formed between 5-FdUMP, thymidilate synthase, and tetrahydrofolate is slowly dissociable with a half-life of 6 hours in intact cells. The half-life of this dissociation may also be different depending upon the time-of-day at which it is formed. A folate cofactor is an absolute requirement for complex formation; severe depletion of intracellular reduced folates compromises complex formation [92]. FUDR catabolism. The importance of FdUMP degrading enzymes in determining cytotoxicity has not been fully explored. FdUMP may be dephosphorylated and converted into FUDR by 5'-nucleotidase and phosphatase enzymes [93]. The formation of fluoro-deoxy-uridine-di-phosphate (FdUMP) and -tri-phosphate (FdUTP) takes place but seems to be quantitatively less important [94]. FUDR is split by the ubiquitous enzyme pyrimidine nucleoside phosphorylase (PNP) into FU and deoxyribose-1-phosphate. It is not currently known whether any of these enzymes are circadian rhythmic in their activities. FU activation. FU may be anabolized in a multi-step process, which results in the formation of its main active metabolite, fluoro-uridine-tri-phosphate (FUTP), interfering with RNA synthesis and to a lesser degree, to FdUMP which then interacts with TS. The degree of activation and catabolism depends upon the regulating enzyme capacity. Activation is promoted by ortate phosphori-bosyl transferase (OPRT), pyrimidine nucleoside phosphorylase and uridine and thymidine kinases [93]. FU catabolism. The major FU-degrading enzyme, dehydropyrimidine dehydrogenase (DPD), reduces FU to dehydrofluorouracil (DHFU). The activity of this enzyme is rate limiting for FU degradation [95]. During subsequent degradation, alpha-fluoro-beta ureido proprionic acid (FUPA), alpha-fluoro-beta guanido proprionic acid (FG-PA), alpha-fluoro-beta alanine (FBAL), urea, carbon dioxide and ammonia are formed. None of these products are cytotoxic. The main organ for fluoropyrimidine metabolism is the liver. There is increasing information that FU catabolism is highly coordinated in circadian time (Table 2). Elimination of FU and FUDR from the plasma is a rapid process. The plasma half-life of FU after IV injection in patients is anywhere between 6 and 20 minutes with considerable inter-individual variation. Most of the drug is converted into carbon dioxide. Less than 20% is excreted in the urine. The conjugation with bile acid has been described [96], which may be of importance for the development of cholestasis and cholangitis after hepatic arterial infusion of fluoro-pyrimidines. Bile acid production has a profound non-meal dependent circadian-stage dependency, leading to the possibility that conjugation dependent drug inactivation is also time-of-day dependent. Influence of drug administration mode, dose and duration on toxicology. The metabolic pathway of FUDR depends on the method of drug administration. The clearance of dose FU, given by a single bolus, is much slower than the clearance of the same dose given over 24 hours, presumably due to saturation of the degradative enzyme DPD [95]. If given in massive amounts over a short period of time as bolus injection, most of it is split into FU and deoxyribose-1-phosphate. However, if given at low dose over prolonged time periods, most of it is "salvaged" and converted into FdUMP inside the cells. This is reflected by a profoundly changing pattern of drug toxicity with increasing infusion duration. The dose limiting side effect for bolus injection is bone marrow toxicity. This might correlate in part with direct FU incorporation into bone marrow cells RNA via FUTP in addition to blockage of DNA synthesis via FdUMP and impaired RNA synthesis via FUTP [97]. For continuous long-term infusion, toxicity along the entire gastrointestinal tract becomes dose limiting with the development of ulcerations and profuse diarrhea. There is no clinically significant bone marrow toxicity when the drug is given by long-term infusion either arterially or intravenously. Part of the explanation for this dramatic difference in toxicity may be the accumulation of FdUMP, which is found at relatively higher levels in epithelial cells of the gastrointestinal tract as compared to bone marrow cells for the same plasma levels of FUDR [98]. Obviously, the circadian time structure of each of the regulating enzyme activities in the anabolism and catabolism of fluoropyrimidines could be important in determining the time dependency of FU and FUDR toxicities seen in listed bolus and infusional preclinical and clinical experiments, eg. DPD described above. Summary. A growing number of fluoropyrimidine studies have addressed the issue of circadian drug timing. They have demonstrated that 5-fluorouracil (FU) is better tolerated during mid-to-late rest span of the recipients' day, and that 5-fluoro-2'-deoxyuridine (FUDR) is less toxic during the daily late activity and early rest spans. The circadian difference in maximum tolerated dose is substantial and frequently exceeds 50% of the mean (i.e., the maximum average dose tolerated at all circadian stages). Drug timing at a stage of reduced toxicity does not diminish anti-tumor activity. On the contrary, tumors seem to be more sensitive at this stage. In addition, because more drug may be given safely, the anti-tumor effect may be even better if a positive dose-response relationship exists for the agent in question. Initial explanations for these observations of circadian stage dependent drug effects come from circadian studies of fluoropyrimidine pharmacokinetics and investigations in cytokinetics for toxicity target tissues. The first careful attempts to clinically utilize our preclinical experience have been successful in reducing toxicity and allowing higher dose intensity. Other chemotherapeutics and combined therapies. Preclinical data. Murine L1210 leukemia has been extensively evaluated at the University of Minnesota and Albany Medical College with regard to the chronobiology of anticancer therapy. A number of studies involving over a thousand mice [69,70,99] demonstrate that a circadian sinusoidal modulation of the 3-hourly doses of cytosine arabinoside can double the cure rate after the inoculation of L1210 leukemia cells and increase the survival time by 60 percent when the highest doses were given near the time of best circadian drug tolerance. A critical evaluation of additional data confirms these findings [100]. Combining agents has been advocated to circumvent drug resistance [101], as demonstrated for L1210 leukemia treated with cytosine arabinoside (ara C) and 1-(2-chlorethyl)-3-cyclohexyl-1- nitrosourea (CCNU) [102], resulting in what has been called "therapeutic synergism" [103]. Using several model systems, we tested one of the drugs present in each of several combination regimens for circadian stage-dependent antitumor effectiveness. Thus, L1210 leukemia was treated with a combination of cytosine arabinoside and cylophosphamide [104], or doxorubicin and cyclophosphamide [105,106], a rat immunocytoma was treated with a combination of doxorubicin and cisplatin [107,108]; and a rat mammary adenocarcinoma was treated with doxorubicin and L-phenylalanine mustard. It was found that an improved therapeutic effect was observed by giving chemotherapy at certain circadian stages even when more than one drug was administered to an animal (Table 7). Many other investigators have demonstrated cytokinetic circadian periodicity in non-human systems [109-117]. It is highly likely that these findings seen in animals are pertinent to the treatment of human beings with anticancer drugs, both in regard to the efficacy of treatment and to the toxicity of the drugs. Glucocorticoids and other hormones may attract the toxicity and the efficacy of antineoplastic agents in either a favorable or unfavorable way. This is particularly relevant since hormones are often administered concurrently with cancer chemotherapy. Moreover, high doses of glucocorticosteroids are often used as anti-emetics, mostly before and after cisplatin administration. Preclinical studies suggest that concurrent high dose methylprednisolone and cisplatin administration may reduce the therapeutic index of the latter [118]. Clearly, further work is needed to define the temporal aspects of drug interactions and the effects on the time structures of the treatment recipient. Clinical experiments. Based upon preclinical studies indicating a better tolerance for VP-16 during the late rest/early awakening phase [119], a randomized, multi-center trial was started in Belgium and France, offering chronotherapy with VP-16 and cisplatin to patients with metastatic non-small cell lung cancer every 4 weeks [120]. Patients have been randomized to receive VP-16 at 100 mg/m2/day on days 1-3 either at 6 or at 18 hours and 100 mg/m2 of cisplatin on day 4 at 18 hours. An interim analysis of 76 cases has not yet demonstrated significant differences in dose intensity or toxicity between the treatment groups. Focan [121] reported on 64 patients who received the same drug combination of methotrexate, 5-fluorouracil, vinblastine, and cyclophosphamide at equal doses but on two different time schedules. Significant differences in tumor responses were found (58% versus 23% partial response rate). Recently, the 5-year disease-free survival of children with acute lymphoblastic leukemia was found to be different, depending upon the timing of their maintenance chemotherapy (6-mercaptopurine + methotrexate); 80% (evening) versus 40% (morning) [122]. Summary. To date only a few metastatic solid tumors can be cured by chemotherapy alone. Most malignancies are either primarily chemotherapy-resistant or will develop resistance. This is especially true for the more frequent cancer types, including those of lung and gastrointestinal origin. While various doses, routes, and modes of drug administration have recently been studied more extensively, less attention has been paid to circadian drug timing and dose intensity. While chemotherapy has reached a plateau of low efficacy in these groups, every potential to make our currently available tools more powerful must be used. Dose intensity and the tumor response are often correlated [58,123]. This indicates the potential importance of any method which can result in a safe increase of chemotherapy doses. Optimal circadian timing of anti-cancer agents offers great promise in this respect. While neither the existence of biological rhythms in all living beings nor the preclinical chronotoxicity study results are seriously questioned, the clinical application of chronochemotherapy is in its infancy, and progress is slow. Several reasons can be cited for the reluctance of many oncologists to study or to adopt the chronotherapy principles, including: 1. The limited understanding of underlying mechanisms chronopharmacodynamic observations: "How does it work?" that explain 2. The complexity of preclinical study results and the apparent difficulty to make simple extrapolations for the design of clinical studies: "What is the best schedule, and how big is the gain?" 3. There is a paucity of convincing clinical data that chronotherapy is better than conventional therapy. However, even well documented chronotherapy (e.g., the time-specified combination of cisplatin with an anthracycline), seems impractical and interfering with routine clinic or hospital schedules: "How can it possibly be done?" The first two issues are inter-related. Our understanding of the malignant processes and the ability of chemotherapy to influence or reverse them is limited. For some of the most frequently used chemotherapeutic agents, the ultimate mechanisms of action are unknown. That makes the explanation of temporal variations even more difficult. However, we can at least identify factors that are likely to play a major role in causing rhythmic changes of drug pharmacodynamics. Chronopharmacokinetics. In the mouse and rat liver, the activity of many enzymes including the cytochrome P-450 system, are circadian stage-dependent, which affects the detoxification capacity for various drugs [124,125]. Circadian differences in blood flow to various organs and renal clearance may also affect drug distribution and elimination [126]. Reproducible circadian differences of pharmacokinetics have been found for cisplatin [48], oxaliplatine [127], carboplatin [128], doxorubicin [129], 6-mercaptopurine, methotrexate [130] and some fluoropyrimidines. Chronocytokinetics. Rhythmic changes in DNA and RNA synthesis, RNA translational activity, and mitotic activity may determine the susceptibility to cell-cycle specific cytotoxins. Such rhythms have been extensively documented in most murine tissues including stomach, duodenum, rectum and bone marrow and used as explanations for the observed rhythms in anti-cancer drug susceptibility [131-134]. Changing the lighting regimen or feeding pattern of animals can predictably shift these rhythms, especially in the intestinum, but not eliminate them [135-136]. Some tumor cells also exhibit circadian-dependent growth while others do not. In general, well-differentiated, slow growing tumors retain a circadian time structure, whereas poorly differentiated, fast growing tumors tend to lose it [137-139]. Circadian-stage dependent cytokinetics of bone marrow cells and the intestinal epithelium may for instance largely determine the susceptibility to fluoropyrimidines. The circadian stages of maximum synthetic activity - and greatest susceptible to an S-phase specific cytotoxic agent - differ between these tissues. In the bone marrow, maximal activity occurs during the early- to mid-rest period [140]; however, it is not known to what extent rhythms in stem cell activity and proliferative activity in the maturing cell compartment are coupled to each other. (Stem cells and progenitor cells should be considered as the most important cells to protect with a "time-shield".) In intestinal epithelium, maximal activity has been described during the late-rest phase of the host [141]. As normal tissues and malignant tissues behave differently in terms of their temporal organization, the circadian timing of optimal therapeutic efficacy and lowest host toxicity frequently overlap. Haus and coworkers [142] have demonstrated with a mouse L1210 leukemia model and Sothern et al. [143] with a rat solid tumor model that the optimal timing of equal dose chemotherapy reproducibly results in a 2-3 fold improvement of tumor cure rate. To date, the toxicities and anti-tumor activities of at least 20 of the most commonly used chemotherapeutic agents have been documented as being circadian stage-dependent both in animals and also for many in humans [144,145]. The difficulty in making simple extrapolations from animal chronotoxicity findings in order to improve clinical treatment schedules are illustrated by the fluoropyrimidine example. FU and FUDR cytotoxicities are clearly circadian stage dependent in animals. However, there does not seem to be one consistently less toxic circadian stage across all studies. For example, the mid-activity optimum for the intestine to tolerate bolus FU is several hours later in the day than is the time optimum for whole animal toxicity and lethality. This observation requires further study with fasted and fed animals in single and multiple dose regimens. It raises, however, the possibility that optimal treatment times may be different for different target tissues as well as for different modes of fluoropyrimidine delivery. In addition, despite the chemical similarity of certain drugs, the circadian time of best drug tolerance may be different. For example, the optimal circadian timing for bolus FU and bolus FUDR is not identical. It is safest to give FU by bolus some time after mid-sleep and prior to usual awakening, while it is safest to give bolus FUDR late in the daily activity span prior to sleep onset. Different metabolic pathways may be responsible for activation of FU and FUDR, and the activity of regulating enzymes may follow different temporal patterns. Chronotoxicity differences have also been found for other closely related drugs (e.g., doxorubicin and epirubicin), while other related drugs (e.g., cisplatin and analogues) have very similar or identical best and worst circadian times for treatment. The different circadian stages for optimal drug timing, depending upon the endpoint studied, may also come from differences in experimental model characteristics (e.g., age, sex, species and strain of the animals, housing conditions, treatment dose, -mode, -route and -schedules, season of experiment performance, etc.). In weighting the data for analysis, one also has to recognize that not all investigators have tested the six circadian stages that are necessary to define a circadian rhythm. Clinical trials testing multiple (i.e., up to six) circadian stages in a randomized fashion are not feasible, so one has to make a best estimate as to what to expect in patients. In spite of the difficulties in extrapolation, clinical studies derived from preclinical experience prove that chronotoxicity also exists in patients, and circadian drug timing can result in less toxicity, which allows for a markedly higher dose-intensity as described above. The ability to reduce toxicity by optimal circadian drug timing, and to safely increase dose intensity, may ultimately translate into improved anti-tumor efficacy, but further confirmation from randomized trials is pending. Our ability to predict an individual's drug susceptibility pattern must also be improved, as many treatment-related variables, recipient related factors, and environmental and seasonal factors may each modulate or shift the optimal circadian treatment time to some extent. To facilitate the practical application of time specified drug delivery schedules, programmable delivery systems have been developed. They are becoming increasingly more powerful tools in terms of their versatility, precision, and reliability. A still relatively small group of researchers and clinical investigators are working to further explore and hopefully answer the major questions that have been raised with chronotherapy. Circadian differences in drug toxicities challenge the concept of standard drug screening and standard clinical phase I/II investigations that consider only one circadian timepoint. These investigations may provide insufficient or misleading information on toxicity, MTD, and anti-tumor activity for untested circadian stages. While the issues remain difficult and complex for the time being, and while no "cookbook recipes" can be offered, undeniable progress has been made. We need to intensify basic research to further explore the mechanisms that determine temporal changes. Clinical phase I/II studies should correlate circadian pharmacokinetics and pharmacodynamics with individual marker rhythms. Randomized multi-center studies should be performed to confirm the current clinical evidence of chronotoxicity. BIOLOGICAL AGENTS. Progress in chronobiology depends on the development of basic knowledge, which usually does not consider the periodicity of the phenomena being examined. Cancer biotherapy is recent, and a chronobiological understanding of this nascent field is rudimentary. Because of the modest amount of relevant work performed to date, this section will focus upon the theoretical basis for anticipation of the circadian organization of the relevant immune networks. Hopefully, the analysis of available data will convince the reader that the response of the organism to the action of the agents to be discussed (TNF, IL-2, interferon, erythropoietin) is, without exception, highly dependent upon the circadian timing of their administration. Cytokines: Tumor Necrosis Factor (TNF) and Interleukin-2 (IL-2). Theoretical basis for time-dependence of cytokine/growth factors related immunological phenomena. Circumstantial evidence supporting the circadian coordination of cytokine/growth factor/blood cells network. The first biochemical circadian rhythm identified in human beings was the daily glucocorticoid pattern, which was discovered by tracing its footsteps, i.e., the circadian pattern of the concentration of certain cellular elements of blood, especially eosinophils [146,147]. Since the concentration of these and all other cells in the blood are profoundly influenced by growth factor availability, which is in turn regulated by monokines and lymphokines, particularly IL-1, these early observations of circadian rhythms in the cellular elements of blood tend to support the likelihood that cytokine biology is under tight circadian coordination. Current understanding of the details of how these cytokine/growth factor/blood cell networks interact is incomplete. There are, however, many pieces of circumstantial evidence indicating that these relationships are tightly coordinated in time with a prominent circadian rhythmicity. IL-2 dependent circadian stimulation of Natural Killer cell production. Important components of cellular immunological response in which TNF and IL-2 play an important role are coordinated in a circadian dependent manner. Lymphokine Activated Killer cells (LAK) and Natural Killer cells (NK) are the two heterogenous lymphocyte populations comprised, at least in part, of nonspecific cytotoxic T lymphocytes produced in response either to an immune challenge, and/or to a subsequent increase in IL-2 activity. These cells are able to lyse a variety of neoplastic and non-neoplastic tissues [148-154]. NK cells, which have been the most well studied, are an important factor inhibiting both the intravascular blood-borne phase of solid tumor metastases [155] and the extravasation and/or early implantation phase of the metastatic process [156]. Well documented circadian rhythms in NK cell activity both in rodents and in humans are established [157-161]. If endogenous IL-2 significantly influences the level of NK activity, this is evidence for either circadian coordination of target lymphocyte IL-2 availability or sensitivity to IL-2. Evidence for Cortisol-IL-1 Circadian Negative Feedback Loop. Glucocorticoids affect IL-1 transcription, translation and cellular release. Non-chronobiologic evidence for the likely chronobiological circadian coordination of IL-1 regulation is mounting. Very recent attempts have been made to explain some of the anti-inflammatory properties of steroids was made. Dexamethasone was shown to inhibit the production and release of IL-1? by human monocytes [162], and soon afterwards glucocorticoids were demonstrated to selectively inhibit the transcription of the IL-1? gene and to decrease the stability of IL-1? mRNA [163]. Since glucocorticoids are exquisitely circadian rhythmic in their production and release, such a profound glucocorticoid effect upon the production of such a central molecule as IL-1 suggests that cytokine/growth factor networks may be tightly coordinated in a circadian manner by the hypothalamo/pituitary/adrenal axis. IL-1 influences cortisol effects. It has been reported that IL-1 decreases cytosolic binding of glucocorticoids in the liver, leading to reduced steroid induction of gluconeogenic enzymes. It has been also found that human recombinant IL-1? affects intracellular glucocorticosteroid binding in rat hepatoma cells. Binding assays revealed that IL-1? significantly reduced dexamethasone binding within 2-4 hours following interleukin administration. Analysis of data indicated that IL-1? treatment decreases the number of available cytosolic receptors. It may, in turn, affect the downstream action of glucocorticoid upon gene expression of IL-1, so closing the loop of negative glucocorticoid/IL-1 interaction. TNF/glucocorticoid interactions at a level of their cytotoxic effects. Physiologic cortisol concentrations influence TNF cytotoxicity. It has been attempted to clarify the regulation of tumor necrosis factor biology by examining the effects of glucocorticoids upon the toxicology of TNF. Purified human recombinant TNF incubated at concentrations of 400 U/mL at 37°C for 40 hours with L929 fibroblasts resulted in 25% survival of these cells. Glucocorticoids added 1 hour before TNF treatment [Hydrocortisone at 1x10-7M (molar concentration) and Dexamethasone at 1x10-7M] substantially inhibited TNF toxicity, resulting in 88% and 89% fibroblast survival, respectively. Normal human plasma contains 1.3-5.5x10-7M of hydrocortisone. Therefore, these data indicate that hydrocortisone may potentially act as an endogenous in vivo modulator of TNF activity, which may, in part, help explain the extremely variable TNF toxicity between patients. Since corticosteroid levels are highly circadian stage dependent, large circadian stage dependent differences in the toxic/therapeutic ratio of this drug can be expected. Further evidence for possible glucocorticoid/cytokine interaction: glucocorticoid mediated cytotoxicity. Cytotoxic T lymphocytes (CTL) induce apoptosis, a cytolytic process in target cells which effects a rapid and characteristic degradation of chromosomal DNA. Glucocorticoid-mediated cytolysis of immature thymocytes resembles this process to a high degree [164]. The possibility has been explored that these two lethal processes share the common pathway by studying the susceptibility of glucocorticoid-resistant mutants to CTL-mediated killing. He reports that an unusual thymoma mutant (having normal glucocorticoid hormone receptor activity) is resistant to both glucocorticoid and CTL. What is more, a single-step genetic reversion can restore sensitivity to both glucocorticoid and CTL. The genetic locus thus identified reveals one element of the endogenous cell suicide pathway that can be triggered by different effectors, corticosterone and CTL. These data, together with those presented in the former paragraph, indicate how the profound circadian dependency of cytokine-induced cytotoxicity of glucocorticoid and/or CTL sensitive cells may, in principle, be directly modulated by an endogenous circadian rhythm in the glucocorticoid concentration. Circadian dependent modulation of glucocorticoid production by IL-2. Sanchez de la Pe?a [165] has attempted to discover whether the interaction between neuroendocrine and immune networks, as mediated by IL-2, is circadian stage dependent light/dark standardized. Hence, rats were killed at one of 6 circadian times of day and quarted adrenals or isolated adrenocortical cells were then incubated for 60 min with or without IL-2. It was found that IL-2 stimulated corticosterone production in adrenal cells harvested from rats at usual daily arousal (p=0.02), but not those harvested at other times of day. Analysis revealed a circadian rhythm in IL-2 effect upon cortisol production (cosinor p=0.014). These results show that the in vitro action of IL-2 upon the adrenal cortex is circadian stage dependent. Role of IL-1 in circadian regulation of body temperature and fever. Early observations by Dinarello raising the possibility that IL-1 may be an important molecular mediator of fever, independently raise the likelihood of tight cytokine circadian control [166]. Febrile reactions secondary to a wide variety of infectious, inflammatory and malignant processes do not occur at random. They almost always occur in the evening, at the time of the day usually associated with the daily nadir of cortisol. In fact no one, as yet, has attempted to explain the molecular basis of the normal daily temperature rhythm. If IL-1 does influence temperature regulation, this prominent circadian rhythm which serves as one of the main circadian markers for chronobiologic studies, peaking near the usual circadian cortisol nadir, may be viewed as circumstantial evidence for temporal cytokine coordination. Extrapolating Dinarello's reasoning, the normal circadian rhythm in body temperature may represent cortisol/IL-1 counterbalancing one another and a resultant temporally coordinated cytokine circadian biology. Cell cycle-specific effects of TNF. Each important target tissue of TNF toxicity including immunocytes, nucleated bone marrow precursors, the gut epithelium and the skin, undergo DNA synthesis and traverse through all other phases of the cell cycle non-randomly (rhythmically) throughout the day. Experiments by Darzynkiewicz [167] demonstrate that either the lophase or some very early postmitotic cell cycle stage appears to be the specific point in the cytokinetic cycle at which TNF treated cells undergo lysis. This conclusion is also supported by earlier observations of cells in culture, indicating that, in the presence of tumor necrosis factor, many cells divide and subsequently lyse. As cell cycle is a process showing a prominent circadian coordinated periodicity these data strongly suggest that TNF cytotoxicity will also undergo strong diurnal rhythmic modulation. Central circadian coordination of cytokine biology. Lotz, et al. [168] have recently demonstrated prominent effects of neuropeptides upon the production of IL-1, TNF ??and IL-6 by monocytes. Two groups of mediators, the neuropeptide substance P and K and the monocyte-derived cytokines interact in the neural regulation of immunological and inflammatory responses. Both substance P and K as well as the carboxyl-terminal peptide SP (4-11) induce the release of the cytokines mentioned above. Since monocyte-derived cytokines regulate multiple cellular functions in inflammation and immunity, and since neuropeptides can be released from peripheral nerve endings into surrounding tissues, these findings identify a potent mechanism for nervous system circadian regulation of host defense responses [168]. All plasma and spinal fluid concentrations of neuropeptides studied chronobiologically participate in high amplitude circadian rhythms which are coordinated through the interaction of the suprachiasmatic nucleus - pineal neurophysis - pituitary - adrenal networks. It would be attractive to think of a circadian IL-1 rhythm balanced in time by cortisol suppression and substance P and/or K induction (see Figure 24). IL-1 - Central molecule in cytokines-growth factors network. Interleukin 1 (? and ?) seem(s) to be in a central position in chronobiologic coordination of cytokine production, release, toxicity and activity. IL-1 shows anticancer activity, TNF-like hemodynamic activity and growth factor activity. IL-1 alpha has in vivo antitumor activity against Meth A sarcoma, colon 26 adenocarcinoma, B16 melanoma and Lewis lung carcinoma [169]. Both IL-1 molecules also induce a shock-like state in rabbits and this state can be synergistically worsened by concurrent TNF administration, indicating an in vivo interaction between these cytokines [170]. As well as having anticancer and pro-inflammatory activity, interleukin-1 alpha and beta also enhance the release of bone marrow neutrophils and activate circulating tissue phase phagocytes. They also increase the bone marrow production of neutrophils and monocytes by stimulating fibroblasts to synthesize the growth factors GM-CSF and G-CSF [171]. It seems as though most cytokines have multiple, complementary and/or synergistic effects with one another. If the regulation of these factors is coordinately arranged anatomically, they are probably also dynamically coordinated in circadian time. The IL-1 dependent IL-2 synthesis. In the process of immunostimulation, the T-cell networks are affected by Interleukin-1. This cytokine, which can be produced by many cell types, but usually the macrophage, must interact with the T-cell as the macrophage is presenting the antigen to which this T-cell will ultimately respond. One of the most critically important responses following this IL-1 dependent initiation of the cellular immune response is the quick (6-9 hour) expression of the IL-2 gene, production of IL-2 mRNA and production of the IL-2 protein. Subsequently and relatively quickly, IL-2 receptor begins to be expressed in high concentrations. These dynamics subsequently result in an amplification of the local and systemic immunocyte response. Experimental evidence for circadian stage dependency of TNF and IL-2 effect. Early experiment with E. coli endotoxin. Indirect evidence for non-trivial reproducible circadian variation in cytokine toxicology was first presented in 1960, when work in Halberg's laboratory connected the chronobiological studies to all of these early and more recent observations. These studies showed large magnitude circadian stage dependence of the lethal toxicities of E. coli endotoxin (Figure 25) [172]. It has been also shown that TNF is an important mediator of endotoxin-induced septic shock. This, interpreted in light of these 1960 findings, would indicate that the lethal and nonlethal toxicities of this and perhaps other cytokines are likely to be very different at different times in the circadian cycles of treated animals. Our recent results with TNF support this hypothesis. TNF studies. The advent of an ever-increasing number of genetically-engineered biological substances having anticancer activity has been tempered by unpredictable and poorly reproducible toxicities. Recombinant human tumor necrosis factor (TNF) has shown both toxicity and anticancer activity in transplantable tumor models [173]. In a series of experiments, we examined the effect of administration timing of TNF on toxicity and therapeutic effectiveness [174,175]. We gave a single IV injection of either 250, 500, 750 or 1000 μg/kg of TNF to 8-10 week old female Balb/C mice and 1000 or 1500 μg/kg to 15-16 week old mice, kept under a 12:12 lights on/off schedule. The 240 mice were time-randomized to receive their injection at one of 6 4-hourly circadian stages and followed for survival. Mortality across the 6 test times was compared by Chi-square analysis with the following results: Less than 50% overall mortality was reached in each of the first four doses. Except for the lowest dose, mice treated during mid to late activity (18-22 HALO) lived longer and died less frequently than those injected during the resting phase. For the highest doses (1000-1500 μg/kg), mice injected during their activity span had less than 30% mortality as compared to 70 to 100% for those treated during the resting phase (02-10 HALO) (Figure 26). The circulatory collapse and shock may represent a final common pathway of endotoxin mediated lethality, and, indeed, the mortality from TNF displays a circadian pattern strikingly similar to that reported in mice from E. coli endotoxin nearly 3 decades ago [172]. In a preliminary study on tumor-bearing mice, 75 female Balb/C mice with Meth A sarcoma were treated daily for seven days, or every other day for 14 days, with 1 μg/mouse. The tumor growth was much slower in animals treated with TNF then in those treated with saline, our control group. The effect of TNF was highly dependent on the circadian stage of its administration (F =2.2, p = 0.07 from analysis of variance). Fewest cures were observed in the daily activity span, just after the daily peak in corticosterone (Figure 27). We concluded that the toxicity of TNF and its antitumor activity was highly circadian-stage dependent and in need of further study to deduce the proper timing to result in its most effective use for clinical success. IL-2 study. In another trial, we investigated dependence of IL-2 effects upon the time of its administration. To achieve this goal we injected 60 female C3H mice with 25,000 U/24h or the same volume of excipient (control) during 3 consecutive days i.p., at 6 circadian stages. Mice were killed 48 hours after the last IL-2 injection, and cell suspensions from spleen and bone marrow were prepared. The endpoints of IL-2 effect were: cell counts and proportion of different lymphocyte subsets, spleen weight and wet-dry lung weight. Analysis of data showed an increase in the number of spleen cells by 16% overall, but up to 3-fold more when IL-2 was given during the activity span of the animals (p<0.02). Spleen weight was also correspondingly dependent upon timing of IL-2 administration. The capacity of this molecule to modify the proportion of lymphocyte subsets was very different, depending upon when in the day it was given, and an increase in number of NK cells by >50% took place only in those animals treated during their daily activity span. Toxicity, as gauged by wet-dry lung weight, was also circadian stage dependent. If the circadian timing of IL-2 and TNF is ignored it can be expected that experimental and clinical results will be highly variable and (apparently) irreproducible. Interferon. Alpha-interferon is active against a variety of malignancies in phase II studies, including leukemia (especially hairy cell leukemia), malignant lymphoma, multiple myeloma, melanoma, colon cancer, renal cell carcinoma, some AIDS-related malignancies, and chronic hepatitis B and C. It is being used both as a monotherapy and in combinations with other biological agents (e.g., IL-2) or classic chemotherapeutic agents. Its optimal mode, schedule and time of day for delivery are unclear. The rather limited efficacy of this drug needs to be improved, likewise serious attempts to diminish interferon side effects, including flu-like symptoms (fever, myalgia, athralgia, malaise, fatigue), nausea, vomiting, diarrhea, anorexia, renal and liver injury, bone marrow suppression, somnolence and mental deterioration which will often force physicians either to diminish the dose of this drug or to stop therapy should be encouraged. Theoretical basis for better efficacy and fewer side effects after early night interferon treatment. Bocci has described a theoretical basis for expecting better efficacy and fewer side effects if night time interferon administration is prescribed [176]. He also gives that there are significant and highly reproducible diurnal variations of several lymphocyte subsets, with the lowest levels of many occurring daily at 8 to 10 hrs in the early part of the diurnal activity span, and the highest daily levels occurring just prior to nocturnal sleep onset [177-180]. Levels of circulating monocytes are also low upon arising and high in the evening. On the other hand, cortisol concentration remains at a low level during most of the night, increases rapidly in the early morning hours, and reaches maximum at about 8 a.m., just at the time of the lowest levels of circulating PBM (peripheral blood mononuclear cells). This negative relationship between cortisol and PBM blood concentration has also been demonstrated in many strains of mice [181]. It is very likely that the early morning increase in plasma cortisol causes a disappearance of PBM, while the decrease in the cortisol level between 8 a.m. and midnight may favor their return into the circulation. On the other hand, plasma levels under physiological conditions are negligible in the morning and tend to increase in the late afternoon [182,183]. Following exogenous administration, IFN peak plasma levels are reached within an hour after IM injection [184]. IFN is removed very rapidly from the blood, thus when it is administered in the morning, its plasma peak coincides with the lowest PBM levels, thereby decreasing interaction of the IFN with effector cells. Finally, interferon administration causes a rise in total serum 11-hydroxycorticosteroids, with a peak some 8 hours after injection [185-187]. So when IFN is given daily, the cortisol level, which ebbs normally during the day, will rise, and thus diminishes the expected diurnal increase of PBM. Interestingly, the in vitro responsiveness of PBM to mitogens varies inversely with the level of plasma cortisol and is maximal between 8 p.m. and 2 a.m. in subjects that usually sleep between 11 pm and 7 am [188,189]. Thus, it appears that IFN administered in the evening should cause a minimal systemic disturbance by reinforcing natural diurnal patterns of biovariability, and by preserving the natural circadian coordinate function of the hypothalamus-pituitaryadrenal axis. Moreover, evening administration will permit maximal IFN plasma concentration to coincide with the usual daily maximal concentration of PBM - its effector cell population. Lymphopenia which often follows interferon injection, would also be minimized or would not take place when IFN is given at the time of the day when the most effective immunological response is anticipated. Clinical report about the advantage of night-time interferon administration. A preliminary communication [190] has reported experience of a group of patients treated with recombinant ?-interferon either at night or during the day. Seventy-five percent of the patients (90% of those treated with a low dose) noticed an improvement in their energy level when they received IFN at night, as compared with that experienced when IFN was given during the day. Many patients taking interferon at night for several months have reported completely normal work tolerance and feelings of well-being, which is a rare finding when IFN is administered in the morning. Both the toxicity and efficacy of interferon are very likely to depend upon the circadian stage of their administration. So far, circadian rhythms of endogenous levels of these molecules have been totally disregarded. As a starting point, it seems appropriate to at least fix daily administration timing of the agent to the time of the daily physiological peak. Because the pharmacological administration of cytokines is able to perturb the normal oscillation of hormonal secretion, it should also be given at times that tend least to alter important circadian rhythms of hormonal secretion and circulating pools of effector cells. Improvement of efficacy is probable if the agent is given at times when effector cells are more readily available for activation. Describing an optimal schedule of time-dependent IFN-delivery, however, is at an embryonic stage and intensive preclinical and clinical research is required to explore more fully the probable advantages offered by optimal chronobiological IFN scheduling. Erythropoietin. Human recombinant erythropoietin, the first commercially available growth factor, is effective in ameliorating the anemia of end stage renal failure and pre-dialysis renal insufficiency. Its utility extends to the treatment of anemias caused by malignancies and chemotherapy, as well as is both certain of the anemias associated with AIDS and to chronic anemias caused by inflammatory disorders. It will also be widely used to decrease transfusion requirements, and to allow preoperative banking of larger amounts of autologous blood prior to elective surgery. Evidence for circadian rhythmicity of erythropoiesis. Erythropoiesis is known to be highly rhythmic on the circadian (24 hour) time scale with respect to serum levels of erythropoietin [191], erythroid progenitor cell numbers, the resultant number of reticulocytes and mature erythrocytes [192], and the susceptibility of red cell precursors to myelotoxic drugs [193]. The biologic and biochemical basis for the coordination of these endogenous hematopoietic rhythms remains to be fully explained. There is increasing evidence for the participation of glucocorticoids in coordinating the circadian rhythm in erythropoiesis. The circadian variation in serum glucocorticoid levels in man and animals is well known. These highly circadian-stage dependent steroids have been shown at physiologic concentrations to promote the survival and proliferation of human erythroid and myeloid precursors in vitro [194, and Chikkappa and Pasquale, unpublished]. The role of these steroids in coordinating the circadian-based hematopoietic rhythms has not been defined. The stimulation of human myeloid precursor cell growth by glucocorticoids in vitro has been shown to be a result of an indirect effect on accessory (non-precursor cell) cells, and can be reproduced by supernatants of such cells [194]. It has been hypothesized that this effect could be due to the production or activation of positive growth-promoting factors (eg. IL-3, GM-CSF), or the withdrawal or inhibition of negative growth factors. Glucocorticoids are known to decrease the production of several cytokines that are inhibitors of erythropoiesis, such as TNF [195] and interferon [196]. This inhibition of cytokine production by glucocorticoids may occur at the level of a 3' UA-rich regulatory region in these mRNAs which regulate their stability and may control translational efficiency of these RNA species [197,198]. Experimental evidence for circadian character of erythropoietin biology. Circadian rhythm of endogenous EPO concentration in human serum. When a sensitive radioimmunoassay for serum erythropoietin level (s-EPO) (using labelled rHuEPO) and antiserum to urinary rEPO were used, Wide, et al. [191] noticed a large intra-individual variation. In order to investigate whether a circadian rhythm in endogenous serum erythropoietin levels was the cause of this variability, they studied serum EPO level in 27 hospitalized adult patients suffering from various illnesses, but all of whom had a level of s-EPO at 8-12hrs within the range of healthy individuals, and had normal cortisol levels during the 24 h. A well-marked circadian rhythm of serum EPO levels was found. Its peak was placed at 20 hrs, and its trough at 8 hrs. Although the mechanisms behind the observed rhythm are unknown, these results confirm a circadian character of erythropoietin biology. Circadian-dependent response of the bone marrow upon EPO administration. Erythropoietic effect of EPO may differ in healthy and anemic organisms as well as in different anemias. To investigate these possibilities, we have used three different animal models: a) intact mice, b) mice made anemic by bleeding, and c) mice anemic as a result of bearing the transplantable tumor. In the course of the studies presented in this chapter, intensity of EPO-induced hematopoiesis was estimated on the basis of hematocrit and reticulocyte determinations. Circadian stage dependent erythropoietin response in intact female CD2F1 mice. Our first EPO trial was performed on 90 intact CD2F1 female mice [199]. They were injected subcutaneously with a single dose of recombinant human erythropoietin (rHuEPO; 300 IU/kg of b.w.) at one of six different circadian times. Erythropoietic response was evaluated by hematocrit and reticulocyte determinations after exsanguination at 24 hour intervals over five days following injection. It was observed that baseline hematocrit and reticulocyte concentrations each varied rhythmically as a function of the circadian stage of measurement. The overall increase in reticulocytes was found to be maximal 48-72 hours after injection. For this single fixed dose of rHuEPO, the rise in reticulocytes varied from a 50% increase when injected at 23 or 1 HALO (early light, beginning of rest in nocturnal animals, usual cortisol low), to a 310% increment when injected at 15 HALO (early dark, beginning of activity) (Figure 28). This time of maximal reticulocyte response roughly follows the known time of maximal corticosterone level in these animals by 4 - 5 hours. Over the 5 days following injection of rHuEPO, the hematocrit response varied from a 9% increase at 72 hours when given at 23 or 1 HALO, to the lack of a significant elevation when given at four other intermediate HALOs (Figure 29). This dissociation of reticulocyte and hematocrit response requires additional study and may relate to the relatively weak effect of a single EPO injection and could also reflect non-hematopoietic changes in hematocrits related to volume status. Erythropoietin injection also resulted in an early (24-48 hours) decrease in hematocrit. The magnitude and timing of this initial rHuEPO-associated hematocrit fall was markedly circadian stage dependent. Circadian stage dependent erythropoietin response in anemic female CD2F1mice. In the next experiment, 60 CD2F1 female mice had been bled for 4 days, so that within this time their HCT levels fell by 10%. The next day, half of the mice were injected with 300IU/kg of EPO, the other half remained untreated and were used as the control group. After erythropoietin administration, hematocrit and reticulocytes were estimated for all animals each day during the 8 following days. Analysis of data revealed peak hematocrit response to the 3 days after EPO administration: it was largest in those mice injected in late rest (10 HALO), and weakest when injected within the activity span (Figure 30). The two-way interaction between EPO effect and time of its injection was significant (p<0.001). Also analysis of reticulocyte response upon rHuEPO injection showed its strong dependence upon timing of drug administration: significant response of young red blood cells appeared during animals' rest period only, with the peak at 10 HALO. The lowest response took place in the mid- to late activity span (Figure 31). The EPO effect and injection/time interaction, however, did not reach statistical significance (p=0.084). Low dose of erythropoietin did not influence murine bone marrow sufficiently to make this interaction significant. Two-way ANOVAs for each HALO, however, showed large differences in EPO effect upon reticulocyte counts. Circadian stage dependent erythropoietin response in anemic, female C3HeB/FeJ mice bearing transplantable mammary carcinoma. Another experiment was performed in order to investigate changes in HCT and RTC levels when EPO was administered at different times of the day in the model of tumor bearing mice. Twenty-five female C3HeB/FeJ mice were inoculated subcutaneously with mammary carcinoma which occurs spontaneously in this strain of mice. Two weeks later, tumor was palpable by 85% of animals, with a corresponding 14% fall in hematocrit (compared with the values on day of tumor innoculation). Fourteen days after injection of malignant cells, EPO was administered to half of the mice (1200 IU/kg s.c.), and an equal volume of diluent to the other half (control). Within the following days, blood was examined for hematocrit and reticulocytes levels. Analysis of data revealed a significant influence of the time of EPO injection upon both hematocrit (p<0.001) and reticulocyte counts (p=0.022). Hematocrit was significantly improved only in those animals injected at 10 HALO (p=0.014). In both other groups of mice (those injected at 2 and 18 HALO), the influence of EPO upon hematocrit was not significant (Figure 32). Levels of reticulocytes were significantly higher only in those animals injected at 18 HALO (r=0.013). Those injected at 2 and 10 HALO did not show any significant change in RTC count. At some circadian stages a lower erythropoietin dose has a greater effect upon hematocrit than higher doses. A further experiment was carried out at two timepoints, i.e., 2 and 18 HALO, in order to test for circadian dependence of the effect of three different doses of recombinant human erythropoietin (300, 600 and 1200 IU per kg of body weight) upon hematocrit levels in CD2F1 female mice. Results showed that the greatest HCT rise in mice injected at 2 HALO appeared three days after administration of the hormone. A dose of 1200 IU/kg turned out to be the most effective in this group. However, in mice injected at 18 HALO, the peak HCT response was observed to be 2 to 4 days after treatment, and was greatest in those animals injected with the lowest dose of EPO. The influence of both time and dose of EPO administration upon hematocrit levels were statistically significant (ANOVA: p<0.001 and p<0.01 respectively). This series of preliminary findings confirms a time dependence for the effect of erythropoietin. The final pattern of proper circadian timing of EPO administration, however, needs further study. Also, its interpretation in the context of such factors as age, stage of estrus cycle, and season of the year has to be performed. If these results can help to describe the most effective schedule for EPO delivery, it could radically reduce the required amounts of EPO, which is very expensive. For example, in the USA, there are about 100,000 patients annually who would benefit from dialysis combined with EPO treatment. Given that basic EPO treatment costs are $5,000 per patient annually, it can be seen that if circadian modulation of drug delivery can reduce the amount of EPO required by 30% (which, on the grounds of our results, seems to be highly possible) then this would result in a potential reduction in cost of EPO by $150 million per annum! The intelligent use of biological therapy. While it has been adequately demonstrated that the therapeutic ratio of toxicity to anticancer drugs is dependent to a large extent upon the timing of drug delivery (i.e., relative to circadian time, time between doses, and drug sequence), these variables are much more relevant to the effective use of biological response modifiers. The time of day when interferons, tumor necrosis factors, IL-2, LAK cells or growth factors are given - relative to the patient's circadian cycle, repeat dosages, their sequences among one another, and standard cytotoxic treatment - ultimately will determine how effective these approaches are. It is clear that the standard Phase I and II approaches to the study of biological agents will not adequately define either their activity or even the relevant toxicities of these agents, even though they have profound biological effects in picogram quantitates while the relationship between host and tumor is elegantly and temporally complex. Administering milligram quantities of biologicals without regard to this complexity can only result in excessive expense, higher toxicities than necessary and finally, great frustration. The studies described above give that the availability of instruments able to stipulate sequence, interval, circadian stage and infradian pattern of immune modulation is a sine qua non to optimal biotherapy. FUTURE PERSPECTIVES OF CHRONOTHERAPY OF CANCER. The suggestion that chemotherapy schedules adjusted to circadian rhythms will result in better drug tolerance and higher efficacy dates almost 20 years, to when Haus et al. [99] reported the circadian stage-dependence of arabinosyl-cytosine tolerance and efficacy in leukemic mice. Since then, experimental evidence documenting the critical importance of circadian timing of cytotoxic treatment has been mounting steadily [200]. The most commonly used anticancer drugs have been shown to be either substantially less toxic, more effective, or both, at certain circadian stages. More recently, genetically engineered biological response modifiers and growth factors have entered clinical trials. They form a broader basis for hope in the fight against the solid tumors that most commonly afflict humans. Initial chronobiologic studies showed that these new therapeutic agents can be several-fold more exquisitely circadian-stage dependent in the amount of toxicity and anticancer activity [184]. Proper circadian timing will, therefore, be of growing, rather than diminishing, importance as we learn to use these agents. An entirely independent, yet related, literature documents the sequence and interval dependence of both conventional chemotherapeutic agents and biological response modifiers. As treatment regimens are becoming more complex, further progress against cancer will depend in part upon a more sophisticated (technological) approach to drug administration. Careful extrapolation from our extensive preclinical investigations has allowed us to design time-qualified clinical treatment schedules for doxorubicin and cisplatin. Optimal drug timing resulted in less toxic, higher dose intensity levels being tolerated safely, with improved tumor control and patient survival. Since then, similar clinical studies have been performed elsewhere, with results confirming and emphasizing the importance of circadian drug timing [202]. As the extent of host-tolerance for each anticancer agent tested in humans differed markedly as a function of its dosing time, the extension of circadian-timed therapy is logical [203]. Since inter-individual differences may determine host chronotolerance, and to some extent tumor susceptibility, circadian monitoring of marker variables must be considered and developed. To date only a few metastatic solid tumors can be cured by chemotherapy alone. Most malignancies are primarily chemotherapy-resistant or will develop resistance. This is especially true for the more frequent cancer types. We argue that more attention must be paid to circadian drug timing, mode, route, drug sequence and intervals, and dose intensity to make the currently available pharmacological tools more effective. Priority setting for future study. There are obviously too many anticancer agents to study each of them thoroughly. Even if that could be accomplished, it is not desirable to put the same effort into a chronobiologic study of each and every agent. Clinical considerations must, therefore, determine which drugs are to be studied with respect to chronobiology. Thus, highest priority must be given to agents that have: 1) known clinical activity, or (based upon screening information obtained in cell lines of human tumors in nude mice or standard murine screening systems) have a high likelihood of being active against the commonest solid tumors; 2) apparent predictable (and hopefully steep) dose intensity/response relationships; 3) a narrow therapeutic index; From this, murine studies on bo lus toxicity and therapeutic efficacy of cyclophosfamide show this drug to be highly indicated. This drug has just about the broadest spectrum of clinical activity of any available anticancer agent. It has severe toxicities and a narrow therapeutic index. Its dose intensity response relationship is clear and steep. It is also used in very high doses for a growing number of indications. There are some studies documenting a circadian rhythm in this drug's therapeutic index, but additional intravenous bolus studies are required to clarify the best circadian stages for its clinical use. Automatic time-specified drug delivery systems. Chronobiology introduces to medicine a new factor - time. It makes us not only think differently about human physiology, pushing us out of old stereotypes that result in treating patients' bodies in the same way when they sleep and when they are active, but also brings with it many technical challenges. For example, in the case of cancer treatment, it might appear rather difficult to deliver multi-agent therapy when each drug should be administered as a long-lasting infusion and each in a different circadian-dependent manner. Fortunately, the renaissance of interest in biologic rhythmicity coincides with a period of unparalleled technological advance. Complex devices with one or multiple reservoirs (each reservoir independently, complexly programmable, with many steps which can also be completely coordinated by the program) can now be used for optimized chronotherapy [204,205]. First and second generation devices are currently available. These work reasonably well, and newer generations of computerized drug delivery systems are becoming available. Most infusional therapy is still monotherapy. However, infusion regimens for metagents will need to be eventually be combined with both one another and with timed bolus treatments, as well as with carefully sequenced applied cytokines and growth factors, etc.. Therefore, further generations of automated drug delivery will be multi-channel devices. Each channel will be independently programmable, allowing for concurrent or sequential administration of drugs. All single or multiple channel devices used to deliver timed bolus and or continuous infusion chemotherapy should have flexible programmability and an internal clock and calendar capability. Although wearable devices are most commonly employed for mono- or multi-agent chemotherapy, implanted programmable pumps also work excellently. Unexpectedly, the costs associated with the use of implanted programmable devices may be lower than the use of wearable devices rented from home-care agencies when the treatment exceeds a certain duration (usually 3-4 months). No implantable system is, however, yet available with more than a single reservoir. The next individual step in circadian optimized cancer therapy is the development of closed-loop systems, which are self-regulated by sensors of circadian time-structures and respond with optimal individual drug timing. It is of practical value to describe these instruments currently available for circadian time-based anti-cancer therapy. The first implantable programmable system is the Synchromed pump (Medtronic Inc., Minneapolis, MN, USA). This system is filled, and then programmed transcutaneously by telemetry after surgical implantation. The system can deliver variable-rate infusion cycles or timed bolus injections either intravenously, intra-arterially, or intra-thecally. The pump reservoir has a maximum usable capacity of 18 mL. This system can administer either a single drug or a mixture of two or more drugs which are compatible both with one-another and with the pump components and catheter. Single channel, wearable devices which can be flexibly programmed to deliver time-shaped infusion are not currently available commercially, though a prototype Autosyringe device used by Levi was designed to his own specifications (personal communication with Dean Kaman, DEKA Laboratories). The Strato Medical Corporation (Beverly, MA, USA) produces the only commercially available single channel device with a clock and calendar. This device may be used to increase and decrease the drug flow over time in a very limited circadian cycle. Ivion (Englewood, CO, USA) has produced a 4-channel, flexibly programmable device that can deliver optimal time-specified treatments for up to 4 drugs. The maximum infusate volume is 30 mL per channel. However, the programming procedure requires some skills, and once programmed, on-site schedule modifications are not possible. Such modifications require the programming of a new micro-chip which stores the information and controls the pump's actions. The device is also fairly bulky compared to other portable pumps. Devices in development which should be able to deliver time-based infusion of at least one agent include the new Parker (Strato Medical, Beverly, MA, USA) micro-pump which has up to 24 steps per program cycle. I-Flow Co. (Irvine, CA, USA) will offer a 4-channel wearable device for poly-chemotherapy. None of these companies currently have a closed loop delivery system available, although several are developing this technology. Thus, in the near future, we will be technically equipped to easily administer time-specified mono- or combination therapy. How these systems will be used. It is interesting to speculate on how the findings of chronobiology will translate into clinical practice. Automatic programmable, implanted, wearable, and bedside systems will have uses in virtually every speciality of medicine and surgery. Initially, open-loop and, subsequently, closed-loop multiple drug systems will pervade medical practice. Modern oncology will demand circadian timing-stipulated, multi-drug regimens, biological therapies, and hybrid chemobiotherapies. Each of these types of therapy will not only stipulate the time of day of each dose, but how many hours or days between repeat cycles, as well as the order in which the agents or patterns of agents are to be used. Using numerous drugs in the treatment of cancer is common, making multiple reservoir systems very useful. Implantable systems will be used for loco-regional therapy, for quasi-continuous systemic cell cycle synchronization, and for longterm immune modulation. These systems will be complemented by the use of extracorporeal devices for short-term therapy in the same patients. Protocol design will no longer be constrained by questions of protocol compliance. National cooperative groups and Cooperative Clinical Oncology Programs (C-COPs) will be able to effectively test and apply complex basic findings to the more immediate benefit of the patient with cancer. Endocrinology will someday rely upon implanted, closed-loop, artificial hypothalmus, pituitary, adrenal, and pancreas technologies. In the interim, many advances will be made possible by simple, closed-loop systems. Right now, ultradian (high frequency) and circadian patterned therapy is essential for treating fertility and growth problems, and it will soon be required to alleviate sleep and emotional disorders. Effective biotherapy will require complex administration of a wide variety of drugs and biologicals. Their application in rheumatology, transplantation, cancer therapy, and treatment of immunodeficiency syndromes (including AIDS) will be expedited by these kinds of delivery systems. Also, in hematology, desferoxamine chelation treatment of iron overload may be improved by automatic delivery systems. Cardiologists and nephrologists treating arrhythmias or hypertension soon will be assisted by closed-loop automatic systems, in which these new technologies will "sense" the events that prompt therapeutic interventions. Even before this, however, programmed, circadian-modified, transcutaneous, or intravenous delivery of nitroglycerine, nitroprusside, dopamine, xylocaine, and other drugs may make clinical sense. Rheumatologists, allergists, and pulmonary doctors may use anti-inflammatory and bronchodilators that are best given after careful consideration of their circadian timing, using automatic programmable devices; while gastroenterologists will use H-2 blocking agents which are bioavailable at that time of day associated with highest basal and stimulated acid secretion (i.e., 2-4 a.m.). Intensive care unit acute care medicine may be the first place where multi-agent therapy will be used in a closed-loop manner, administering diverse drugs to patients with multiple organ failure, according to the physiologic - biochemical signals fed to a device directly from the patients' own monitoring systems. These and many other applications will result as these devices mature, become easier to use, and are expanded to do more, more simply. What should the use of these devices mean for the practice of medicine, and how will their use affect chronobiology? The adoption of "intelligent" automatic, programmable, drug delivery devices will make medicine both intrinsically more complex and extrinsically more simple. It will complex drug and biological regimens safer, less error-prone, and less expensive. The therapy of serious diseases will be more uniform. Protocols developed in university centers and by clinical research groups will be widely available to all medical practitioners and, once instituted, will be carried out more exactly. Better medicine will ultimately be more widely available to more citizens. The clinical research to establish these protocols should be funded by collaboration between government, industry, and academics. The pressure to produce and to market new drugs may decrease and might well be replaced by a more rational pressure to discover and effectively protect the best way of giving drugs. A wide variety of devices and competing protocols will keep each system and protocol cost-effective, and this competition will both assure a return on investment and hold down costs. This cost-accounting of clinical applications research will result in less expensive medical care. Finally, the adoption of programmed automatic drug delivery will bring attention to temporal chronobiologic questions which have been, until now, unanswerable. This attention will turn chronobiology into what it truly is - a multidimensional and dynamic perspective on life science. This movement toward considering the temporal aspects of drug action (and administration) will lead to chronobiology becoming an integral part of the make-up of biologists and physicians alike. REFERENCES. 1. Halberg, F., Haus, E., Cardoso, S.S., Scheving, L.E., Kuhl, J.F.W., Shiotsuka, R., Rosene, G., Pauly, J.E., Runge, W., Spalding, J.F., Lee, J., Good, R.A. 1972. Toward a chronotherapy of neoplasia: Tolerance of treatment depends upon host rhythms. Experientia 29,909-934. 2. Scheving, L.E. 1982. Circadian rhythms in cell proliferation: Their importance when investigating the basic mechanism of normal versus abnormal growth. In: 11th Intl Congress of Anatomy, Biological Rhythms in Structure and Function. New York: Alan R.Liss, pp 39-79. 3. Burns, R.E. Circadian rhythmicity in DNA synthesis in untreated mice as a basis for improved chemotherapy. Cancer Res 41,2795-2802, 1981. 4. Mauer, A.M. Dirunal variation of proliferative activity in the human bone marrow. Blood 26,1-7, 1965. 5. Thorud, E., Clausen, O.P.F., Bjerknes, R., et al. The stathmokinetic method of in vivo time-response with special reference to circadian variations in epidermal cell proliferation in the hairless mouse. Cell Tissue Kinet 13,625-634, 1980. 6. Durie, B.G.M, Salmon, S.E., Russell, D.H. 1977. Polyamines as markers of response and disease activity in cancer chemotherapy. Cancer Res 36,214-221. 7. Janne, J., Poso, H., Raina, A. 1978. Polyamines in rapid growth and cancer. Biochem Biophys Acta 473,241-243. 8. Hrushesky, W., Merdink, J., Abdel-Monem, M. 1983. Circadian rhythmicity characterizes monacetyl polyamine urinary excretion. Cancer Res 43,3944-3947. 9. Brown, H.E., Dougherty, T.F. 1956. The diurnal variations of blood leucocytes in normal and adrenalectomized mice. Endocrinology 58,365-375. 10. Kanabrocki, E.L., Scheving, L.E., Halberg, F., et al. 1975. Circadian varition in presumably healthy young soldiers. Department of the Army, Document PF228437, National Technical Information Service, US Department of Commerce, PO Box 1553, Springfield, Virginia. 11. Cove-Smith, J.R., Pownall, R., Kabler, T.A., et al. 1979. Chronopharmacology. In: A Reinberg, G Halbert (eds), International Congress of Pharmacology. Oxford: Pergamon Press, pp 369-373. 12. Tavadia, H., Fleming, K., Hume, P., et al. 1975. Circadian rhythmicity of human plasma cortisol and PHA-induced lymphocyte transformation. Clin ExpImmunol 22,190-193. 13. Fernandes, G., Halberg, F., Yunis, E., et al. 1976. Circadian rhythmic plaque-forming cell response of spleens from mice immunized with SRBC. J Immunol 117,962-966. 14. Fernandes, G., Carandente, F., Halberg, E., et al. 1979. Circadian rhythm in activity of lympholytic natural killer cells from spleens of Fisher rats. J Immunol 123,622-625. 15. Kohler, W.C., Karacan, I., Rennert, O.M. 1972. Circadian variation of RNA in human leukocytes. Nature 238,94-96. 16. Hrushesky, W.J.M., Dell, I., Eaton, J., Halberg, F. 1982. Circadian-stage-dependent effect of doxorubicin upon reduced glutathione in the murine heart. Proc AACR 23,12. 17. Hrushesky, W.J.M. 1983. The clinical application of chronobiology to oncology. Amer J Anat 168:,519-542. 18. Scheving, L.E. 1984. Chronobiology of cell proliferation in mammals: Implications for basic research and cancer chemotherapy. In: Cell Cycle Clocks, LN Edmunds (ed), New York: Marcel-Dekker, pp 455-499. 19. Mayersbach, H.v. 1978. Die zeistruktur des organismus. Auswirkungen auf zellulaere leistungsfaehigkeit und medikamentenempfindlichkeit. Arznisem Forsch/Drug Res 28,1824-1836. 20. Kinlaw, W.B., Fish, L.H., Schwartz, H.L., Oppenheimer, J.H. 1987. Dirunal variation in hepatic expression of the rat S14 gene is synchronized by the photoperiod. Endocrinology 120:1563-1567. 21. Petit, E., Milano, G., Levi, F., Thyss, A., Bailleul, F., Schneider, M. 1987. Circadian rhythm in 5-FU pharmacokinetics during 5-day continuous infusion. Satellite Symposia Proc ECCO 4,293. 22. Levi, F., Hrushesky, W.J.M., Borch, R.F., Pleasants, M.E., Kennedy, B.J., Halberg, F. 1982. Cisplatin urinary pharmacokinetics and nephrotoxicity: A common circadian mechanism. Cancer Treat Rep, 66,1933-1938. 23. Hrushesky, W.J.M., Borch, R., Levi, F. 1982. Circadian time dependence of cisplatin urinary kinetics. Clin Pharmacol Ther 32:330-339. 24. Boughattas, A.N., Levi, F., Roulon, A., Mechkouri, M., Lemaigre, G., Cal, J.C., Camber, J., Reinberg, A., Mathe, G. 1987. Similar circadian rhythm in murine host tolerance for two platinum analogs: Carboplatin (CBDCA) and oxaliplatine (I-OHP). Proc AACR 28,1788. 25. Aherne, G.W., English, J., Burton, N., Arendt, J., Marks, V. 1987. Chronopharmacokinetics and their relationship to toxicity and effect with reference to methotrexate, 6-mercaptopurine and morphine. Satellite Symposia Proc ECCO 4,40. 26. Roemeling, R.v., unpublished results. 27. Nash, R.E., Echave Llanos, J.M. 1971. Circadian variations in DNA synthesis of a fast-growing and a slow-growing hepatoma: DNA synthesis rhythm in hepatoma. J Natl Cancer Inst 47,1007-1012. 28. Burns, E.R., Scheving, L.E., Tsai, T.H. 1979. Circadian rhythms in DNA synthesis and mitosis in normal mice and in mice bearing the Lewis lung carcinoma. Eur J Cancer 15,233-242. 29. Carter, S.K. 1975. Adriamycin: A review. J Natl Cancer Inst 55:1265-1267. 30. Blum, R.H., Carter, S.K. 1974. Adriamycin, a new anticancer drug with significant clinical activity. Ann Intern Med 80:249-259. 31. Von Hoff, D.D., Layard, M.W., Basa, P., Davis, H., Von Hoff, A.L., Rozencweig, M., Muggia, F.M. 1979. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med 91,710-717. 32. Sobrero, A., Muzzulini, C., D'Amore, F., Bogliolo, G., Massa, G., Ghio, R., Pannacciulli, I. 1982. Activity of 4'-epi-doxorubicin on normal hematopoietic precursor cells in mice. Cancer Treat Rep 66,2061-2069. 33. Pearlman, L.F., Chuang, R.Y., Israel, M., Simpkins, H. 1986. Interaction of three second generation anthracyclines with polynucleotides, RNA, DNA and nucleosomes. Cancer Res 46,341-346. 34. Hill, B.T., Whelan, R.D.H. 1982. A comparison of the lethal and kinetic effects of doxorubicin and 4'-epi-doxorubicin in vitro. Tumori 68,29-37. 35. Weenen, H., Van Maanen, J.M.S., DePlanque, M.M. 1984. Metabolism of 4'-modified analogs of doxorubicin. Unique glucoronidation pathway for 4'-epidoxorubicin. Eur J Cancer Clin Oncol 20,919-927. 36. Weenen, H., Lankelma, J., Penders, P.G.M., McVie, J.G., Ten Bokkel, W.W., DePlanque, M.M., Pinedo, H.M. 1983. Pharmacokinetics of 4'-epi-doxorubicin in man. Invest New Drugs 1,59-64. 37. Good, R.A., Sothern, R.B., Stoney, P.J., Simpson, H.W., Halberg, E., Halberg, F. 1977. Circadian state dependence of adriamycin-induced tumor regression and recurrence rates in immunocytoma-bearing LOU rats. Chronobiologia 4,174. 38. Halberg, F., Gupta, B.D., Haus, E., Halberg, E., Deka, A.C., Nelson, W., Sothern, R.B., Cornelissen, G., Lee, J.K., Lakatua, D.J., Scheving, L.E., Burns, E.R. 1977. Steps toward a cancer chronopolytherapy. In: Proc XIVth Intl Cong of Therapeutics, Montepellier, France, L'Expansion Scientifique Francaise, Paris, pp 151-196. 39. Sothern, R.B., Nelson, W.L., Halberg, F. 1977. A circadian rhythm in susceptibility of mice to the anticancer drug, adriamycin. In: Proc XII Intl Conf, Intl Soc for Chronobiology, Washington D.C., Il Ponte, Milano, Italy, pp 433-438. 40. Sothern, R.B., Halberg, F., Good, R.A., Simpson, H.W., Grage, T.B. 1981. Difference in timing of circadian susceptibility rhythm in murine tolerance of chemically-related antimalignant antibiotics: adriamycin and daunomycin. In: CA Walker, CM Winget KFA Soliman (eds), Chronopharmacology and Chronotherapeutics, Tallahassee: Florida A & M University Foundation, pp 257-268. 41. Mormont, M.C., Roemeling, R.v., Sothern, R.B., Berestka, J.S., Langevin, T.R., Olshefski, R., Wick, M., Hrushesky, W.J.M. 1988. Circadian rhythm and seasonal dependence in tolerance of mice to 4'-epi-doxorubicin. Invest New Drugs 6,273-283. 42. Levi, F., Mechkouri, M., Roulon, A., Bailleul, F., Lemaigre, G., Reinberg, A., Mathe, G. 1985. Circadian rhythm in tolerance of mice for the new anthracycline analog 4'-tetrahydropyranyladriamycin (THP). Eur J Cancer & Clin Oncol 121,1245-1251. 43. Levi, F., Blazsek, I., Ferle-Vidovic, A. 1988. Circadian and seasonal rhythm in murine bone marrow colony forming cells affect tolerance for the anticancer agent 4'-tetrahydropyranyladriamycin (THP). Exp Hematol 16,696-701. 44. Mangioni, C., Bolis, G., Pecorelli, S., Bragman, K., Epis, A., Favalli, G., Gambino, A., Landoni, F., Presti, M., Torri, W., Vassena, L., Zanaboni, F., Marsoni, S. 1989. Randomized trial in advanced ovarian cancer comparing cisplatin and carboplatin. J Natl Cancer Inst 81,1464-1471. 45. Kr?mer, H.P., Hoffman, D., Gronau, T., Kolar, C., Sedlacek, H.H. 1989. B-850040: A new cisplatin derivative. Proc AACR 30,462. 46. Levi, F., Hrushesky, W.J.M., Borch, R.F., Pleasants, M.E., Kennedy, B.J., Halberg, F. 1982. Cisplatin urinary pharmacokinetics and nephrotoxicity: A common circadian mechanism. Cancer Treat Rep 66,1933-1938. 47. Hrushesky, W.J.M., Borch, R., Levi, F. 1983. Circadian time dependence of cisplatin urinary kinetics. Clin Pharmacol Therap 32,330-339. 48. Hecquet, B.J., Menadier, J., Bonneterre, J., Adenis, L., Demaille, A. 1985. Time dependence in plasma protein binding of cisplatin. Cancer Treat Rep 69,79-82. 49. Boughattas, N.A., Levi, F., Fournier, C., Lemaigre, F., Roulon, A., Hecquet, B., Mathe, G., Reinberg, A. 1989. Circadian rhythm in toxicities and tissue uptake of 1,2-diammino-cyclohexane (trans-1) oxalatoplatinum (II) in mice. Cancer Res 49,3362-3368. 50. Fraval, H.N.A., Roberts, J.J. 1979. G1 phase Chinese hamster V79-379A cells are inherently more sensitive to platinum bound to their DNA than mid S phase or asynchronously treated cells. Biochem Pharmacol 28,1575-1580. 51. Hrushesky, W.J.M., Levi, F., Halberg, F., et al. 1982. Circadian stage dependence of cis-diamminedichloroplatinum lethal toxicity in rats. Cancer Res 42,945-949. 52. Levi, F., Hrushesky, W.J.M., Blomquist, C.H., et al. 1982. Reduction of cis-diamminedichloroplatinum nephrotoxicity in rats by optimal circaian drug timing. Cancer Res 42,950-955. 53. Levi, F.A., Hrushesky, W.J.M., Halberg, F., et al. 1982. Lethal nephrotoxicity and hematologic toxicity of cis-diamminedichloroplatinum ameliorated by optimal circadian timing and hydration. Eur J Cancer Clin Oncol 18:471-477. 54. Wesson, L.G. 1964. Electrolyte excretion in relation to diurnal cycles of renal function. Medicine 43:547-592. 55. Sothern, R.B., Halberg, F., Halberg, E., Zinneman, H.H., Kennedy, B.J. 1981. Circadian and methodologic aspects of toxicity from cis-diamminedichloroplatinum, adriamycin and methylprednisolone interaction in rats with immunocytoma. In: CA Walker, CM Winget, KFA Soliman (eds), Chronopharmacology and Chronotherapeutics, Tallahassee: Florida A & M University Foundation, pp 247-256. 56. Hrushesky, W.J.M., Haus, E., Lakatua, D.J., Halberg, F., Langevin, T., Kennedy, B.J. 1985. Marker rhythms for cancer chrono-chemotherapy. In: E Haus, HF Kabat (eds), Chronobiology 1981-1983, New York: Karger, p 493-499. 57. Hermida Dominguez, R.C., Sothern, R.B., Halberg, F., Langevin, T.R. 1986. Variability of circadian acrophase of urinary potassium excretion as a potential marker for cancer chronotherapy. In: F Halberg L Reale, B Tarquini (eds), Proc 2nd Intl Conf Medico-Social Aspects of Chronobiology, Instituto Italiano di Medicina Sociale, Rome, pp 313-325. 58. Levin, L., Hryniuk, W. 1987. dose intensity analysis of chemotherapy regimens in ovarian cancer. J Clin Oncol 5,756-767. 59. Young, R.C., Knapp, R.C., Fuks, Z., Disaia, P.J. 1984. Cancer of the ovary. In: VT DeVita, jr., S Hellman, SA Rosenberg (eds), Cancer Principles and Practice of Oncology. Philadelphia: J.P. Lippincott Co., pp 1083-1118. 60. Roemeling, R.v., Hrushesky, W.J.M. 1986. Advanced transitional cell bladder cancer: A treatable disease. Sem Surg Oncol 2,76-89. 61. Roemeling, R.v., Christiansen, N.P., Hrushesky, W.J.M. 1985. Lack of antiemetic effect of high dose metoclopramide. J Clin Oncol 3,1273-1276. 62. Hrushesky, W.J.M. 1985. Circadian timing of cancer chemotherapy. Science 228:73-75. 63. Levin, L., Hryniuk, W. 1987. The use of dose intensity (DI) to solve problemsin gynecologic oncology. Proc ASCO 6,119. 64. Roemeling, R.v., Hrushesky, W.J.M., Fraley, E. 1987. Long-term control of locally advanced transitional cell bladder cancer (TCCB) by high-dose intensity, circadian-based adjuvant chemotherapy. In: SE Salmon (ed), Adjuvant Treatment of Cancer V, Orlando: Grune Stratton, pp 571-580. 65. Levi, F., Benavides, M., Chevelle, C., LeSaunier, F., Bailleul, F., Misset, J.L., Regensberg, C., Vannetzel, J.M., Reinberg, A., Mathe, G, 1990. Chemotherapy of advanced ovarian cancer with 4'-0-tetrahydropyranyl doxorubicin and cisplatin: A randomized phase II trial with an evaluation of circadian timing and dose intensity. J Clin Oncol 8,705-714. 66. Levi, F. (In press.) Chronpharmacology of anticancer agents and cancer chronotherapy. In: H Kummerle (ed), Internaitonal Handbook of Clinical Pharmacology. West Germany: Ecomed, Landsberg am Lech. 67. Halberg, F., Levi, F., Hrusheksy, W., et al. (In press.) Murine chronotolerance of cisdiamminedichloro platinum. II: Circadian rhythm and circannual influence. Proc XIVth Conf Intl Soc Chronobiol. 68. Edmunds, L.N. 1978. clocked cell cycle clocks; Implications toward chronopharmacology and aging. In: Samis HV, Jr., Capoblanco S (eds): Aging and Biological Rhythms. New York: Plenum Publishing, pp 125-184. 69. Halberg, F. 1964. Medical aspects of stress in the military climate. In: Walter Reed Army Institute of Research Symposium, Washington D.C., US Government Printing Office, pp 1-36. 70. Halberg, F., Haus, E., Cardoso, S.S., et al. 1973. Toward a chronotherapy of neoplasia: Tolerance of treatment depends upon host rhythms. Experientia 29,909-1044. 71. Harris, B.E., Song, R., He, Y.-J., Soong, S.-J., Diasio, R.B. 1988. Circadian rhyhtm of rat liver dihydropyrimidine dehydrogenase. Possible relevance to fluoropyrimidine chemotherapy. Biochem Pharamcol 37,4759-4762. 72. Harris, B.E., Song, R., Soong, S.J., Diasio, R.B. 1989. Circadian variation of 5-fluorouracil catabolism in isolated perfused rat liver. Cancer Res 49,6610-6614. 73. Danhauser, L.L., Rustum, Y.M. 1979. A method for continuous drug infusion in unrestrained rats: Its application in evaluating the toxicity of 5-fluorouracil/thymidine combinations. J Lab Clin Med 93,1047-1053. 74. Tuchman, M., Roemeling, R.v., Lanning, R., Sothern, R.B., Hrushesky, W.J.M, 1988. Source of variaiblity of dihydropyrimidine dehydrogenase (DPD) activity in human blood mononuclear cells. In: A Reinberg, M Smolensky, G Labrecque (eds), Annual Review of Chronopharmacology, Vol 5, New York: Pergamon Press, pp 399-402. 75. Harris, B.E., Song, R., Diasio, R.B. 1989. Circadian variation (CV) of dihydropyrimidine dehydrogenase (DPD) activity and plasma 5-fluorouracil (FUra) levels in cancer patients receiving FUra by protracted continuous infusion. Proc AACR 30,247. 76. Petit, E., Milano, G., Levi, F., Thyss, A., Bailleul, F., Schneider, M. 1988. Circadian rhythm-varying plasma concentration of 5-fluorouracil during a five-day continuous venous infusion at a constant rate in cancer patient. Cancer Res 48,1676-1679. 77. Burns, E.R., Beland, S.S. 1984. Effect of biological time on the determination of the LD50 of 5-fluorouracil in mice. Pharmacol 28,296-300. 78. Popovic, P., Popovic, V., Baughman, J. 1982. Circadian rhythm and 5-fluorouracil toxicity in C3H mice. Biomed Therm 25,185-187. 79. Gonzalez, J.L., Sothern, R.B., Thatcher, G., Nguyen, N., Hrushesky, W.J.M. 1989. Substantial difference in timing of murine circadian susceptibility to 5-fluorouracil and FUDR. Proc AACR 30,616. 80. Peters, G.J., Van Dijk, J., Nadal, J.C., Van Groeningen, C.J., Lankelma, J., Pinedo, H.M. 1987. Diurnal variation in the therapeutic efficacy of 5-fluorouracil against murine colon cancer. In Vivo 1,113-118. 81. Gardner, M.L.G., Plumb, J.A. 1981. Diurnal variation in the intestinal toxicity of 5-fluorouracil in the rat. Clin Sci 61,717-722. 82. Roemeling, R.v., Hrushesky, W.J.M. 1987. Circadian pattern of continuous FUDR infusion reduces toxicity. In: Pauly JE, Scheving LE (eds), Advances in Chronobiology, Part B, Allen R.Liss, New York, pp 357-373. 83. Roemeling, R.v., Hrushesky, W.J.M. 1990. Circadian FUDR infusion pattern determines its therapeutic index. J Natl Cancer Inst 82,386-393. 84. Levi, F., Soussan, A., Adam, R., Caussanel, J.P., Metzger, G., Misset, J.L., Descorps-Decleres., Kustlinger, F., Lorphelin, D., Jasmin, C., Bismuth, H., Reinberg, A., Mathe, G. 1989. Programmable-in-time pumps for chronotherapy of patients (pts) with colorectal cancer with 5-day circadian modulated venous infusion of 5-fluorouracil (CVI-5FUra). Proc ASCO 8:111 (abst.). 85. Levi, F. 1989. Ambulatory chronotherapy of colorectal cancer with 5-fluorouracil, folinic acid and oxaliplatinum via a multichannel programmable pump. Results of a phase II trial. Proc Eur Conf Clin Oncol (ECCO) 5, London UK (abst.). 86. von Roemeling, R., Hrushesky, W.J.M. 1989. Circadian patterning of continuous FUDR infusion reduces toxicity and allows higher dose intensity. J Clin Oncol 7,1710-1719. 87. Vugrin, D. 1988. Systemic therapy of metastatic renal cell carcinoma. Semin Nephrol 7,155-162. 88. Wesen, C., Roemeling, R.v., Lanning, R., Grage, T., Olson, G., Hrushesky, W.J.M. (In press.) The effect of circadian modification of intra-arterial FUDR infusion rate upon hepatic toxicity. J Clin Oncol. 89. Chabner, B.A. 1982. Pyrimidine antagonists. In: BA Chabner (ed), Pharmacologic Principles of Cancer Treatment, Philadelphia, W.B. Saunders Co., pp 183-212. 90. Reichard, P., Skold, O., Klein, G. 1959. Possible enzymatic mechanisms for development of resistance against fluorouracil in ascites tumors. Nature 183,939-941. 91. Klubes, P., Connelly, K., Cerna, I. 1978. Effects of 5-fluorodeoxyuridine 5'-monophosphate and 2-deoxyuridine 5'-monophosphate pools and DNA synthesis in solid mous L1210 and rat Walker 256 tumors. Cancer Es 38,2325-2331. 92. Evans, R.M., Laskin, J.D., Hakala, M.T. 1981. Effect of excess folates and deoyinosine on the activity and site of action of 5-fluorouracil. Cancer Res 41,3288. 93. Peters, G.J., Laurensse, E., Leyva, A., Lankelma, J., Pinedo, H.M. 1986. Sensitivity to human, murine and rat cells to 5-fluorouracil and 5'-deoxy-5-fluorouridine in rleation to drug-metabolizing enzymes. Cancer Res 46,20-28. 94. Caradonna, S.J., Cheng, Y.C. 1980. The role of deoxyuridine triphosphate nucleotidohydrolase, uracil DNA glycosylase and DNA polymerase alpha in the metabolism of FUDR in human tumor cells. Molec Pharm 18,513-520. 95. Tuchman, M., Stoeckeler, J.S., Kiang, D.T., O'Dea, R.F., Ramnaraine, M.L., Mirkin, B.L. 1985. Familial pyrimidinemia and pyrimidinuria associated with severe fluorouracil toxicity. New Engl J Med 313,245-249. 96. Sweeny, D.J., Barnes, S., Heggie, G.D., Diasio, R.B. 1987. Metabolism of 5-fluorouracil to anN-cholyl-2-fluoro-?-alanine conjugate: Previously unrecognized role for bile acids in drug conjugation. Proc Natl Acad Sci 84,5439-5443. 97. Schuetz, J.D., Wallace, H.J., Diasio, R.B. 1984. 5-fluorouracil incorporation into DNA of CF-1 mouse bone marrow cells as a possible mechanism of toxicity. Cancer Res 44,1358-1363. 98. Fraile, R.J., Baker, L.H., Buroker, T.R, 1980. Pharmacokinetics of 5-fluorouracil administered orally by rapid intravenous an by slow infusion. Cancer Res 40,2223-2228. 99. Haus, E., Halberg, F., Scheving, L.E., et al. 1972. Increased tolerance of leukemic mice to arabinosyl cytosine with schedule adjusted to circadian system. Science 177,80-82. 100. Halberg, F., Nelson, W., Cornelissen, G., et al. 1979. On methods for testing and achieving cancer chronotherapy. Cancer Treat Rep 63,1428-1430. 101. Goldin, A., Venditti, J.M., Mantel, N. 1974. Combination chemotherapy: Basic considerations. In: Sartorelli AC, Johns DG (eds), Antineoplastic and Immunosuppressive Agents. Berlin/New York: Springer-Verlag, pp 411-448. 102. Scheving, L.E., Haus, E., Kuhl, J.F.W., et al. 1976. Close reproduductio by different laboratories of characteristics of circadian rhythm in 1-?-D-arabinofuranosylcystosine tolerance by mice. Cancer Res 36,1133-1137. 103. Scheving, L.E., Burns, E.R., Pauly, J.E., et al. 1977. Survival and care of leukemic mice after circadian optimization of treatment with cyclophosphamide and 1-?-D-arabinofuranosylcytosine. Cancer Res 37,3648-3655. 104. Tavadia, H., Fleming, K.A., Hume, P., et al. 1977. Circadian variations in the quantity and quality of lymphocyte traffic in human peripheral venous blood. In: McGovern J, Reinberg A, Smolensky MH (eds): Chronobiology in Allergy and Immunology. Springfield, IL, Charles C. Thomas, pp 186-203. 105. Skipper, H.E., Schabel, F.M., Jr., Wilcox, W.S. 1967. Experimental evaluation of potential anticancer agents. XXI: Schedulign of arabinosylcytosine to take advantage of its S phase specificity against leukemia cells. Cancer Chemother Rep 51,125-166. 106. Scheving, L.E. 1981. Eleventh International Congress of Anatomy: Biological Rhythms in Structure and Function. New York: Alan R.Liss, Inc., pp 39-79. 107. Scheving, L.A., Yeh, Y.C., Tsai, T., et al. 1980. Circadian phase-dependent stimulatory effects of epidermal growth factor on deoxyribonucleic acid synthesis in the duodenum, jejunum, ileum, caecum, colon and rectum of the adult male mouse. Endocrinology 106,1498-1503. 108. Brockman, R.W. 1974. Pharmacological basis of cancer chemotherapy. In: Proceedings of the 27th Symposium of Fundamental Cancer Research, M.D. Anderson Hospital and Tumor Institute, Baltimore, Williams and Wilkins, pp 691-771. 109. Scheving, L.A., Yeh, Y.C., Tsai, T.H., et al. 1979. Circadian phase-dependent stimulatory effects of epidermal growth factor on deoxyribonucleic acid synthesis in the tongue, esophagus and stomach of the adult male mouse. Endocrinology 105,1475-1480. 110. Halberg, F., Haus, E., Scheving, L.E. 1978. Sampling of biologic rhythms, chronocytokinetics and experimental oncology. In: Valleron AJ, MacDonald PDM (eds): Biomathematics and Cell Kinetics. Elsevier: North Holland Biomedical Press, pp 175-190. 111. Laerum, O.D., Aardal, N.P. 1981. Chronobiological aspects of bone marrow and blood cells. In: Acosta EV, Mayersbach H, Scheving LE, et al (eds): Eleventh International Congress of Anatomy: Biological Rhythms in Structure and Function. New York: Alan R. Liss, Inc., pp 87-97. 112. Clausen, O.P.F., Thorud, E., Bjerknes, R., et al. 1979. Circadian rhythms in mouse epidermal basal cell proliferation. Variations in compartment size, flux, and phase duration. Cell Tissue Kinet 12,319-337. 113. Rubin, N.H. 1982. Influence of the circadian rhythm in cell division on radiation-induced mitotic delay in vivo. Radiation Res 89,65-76. 114. Mamontov, SG. 1968. Diurnal rhythm of mitoses in the epithelium of the mouse tongue. Bull Exp Biol Med 66,1277-1278. 115. Burns, E.R. 1981. A critique of the practice of comparing control data obtained at a single time point to experimental data obtained at multiple time points. Cell Tissue Kinet 14,219-224. 116. Burns, E.R. 1982. A critique of the practice of plotting data obtained in two in vivo on an 'hours after treatment' format. Oncology 39,250-254. 117. Izquierdo, J.N., Gibbs, S.J. 1974. Turnover of cell-renewing populations undergoing circadian rhythms in cell proliferation. Cell Tissue Kinet 7,99-111. 118. Kodama, M., Kodama, T. 1982. Influence of corticosteroid hormones on the therapeutic efficacy of cyclophosphamide. Gann 73,661-666. 119. Levi, F., Mechkouri, M., Roulon, A., Bailleul, F., Horvath, C., Reinberg, A., Mathe, G. 1985. Circadian rhythm in tolerance of mice for etoposide. Cancer Treat Rep 69,1443-1445. 120. Focan, C., Driesschaert, P., Bastens, B., Mendez, P., Levi, F., Pujol, J.L., Thyss, A. 1990. Circadian tolerance to etoposide and cisplatin as first line treatment for advanced lung carcinoma. Proc IV Intl Conf Chronopharmacol Chronotherapy V-6, Nice France. 121. Focan, C. 1979. Sequential chemotherapy and circadian rhythm in human solid tumors. Cancer Chemother Pharmcol 3,197-202. 122. Rivard, G., Infante-Rivard, C., Hoyeux, C., Champagne, J. 1985. Maintenance chemotherapy for childhood acute lymphoblastic leukemia: Better in the evening. Lancet 2,1264-1266. 123. Hryniuk, W.M., Figueredo, A., Goodyear, M. 1987. Applications of dose intensity to problems in chemotherapy of breast and colorectal cancer. Semin Oncol 14(Suppl 4),3-11. 124. Belanger, P.M. 1988. Chronobiological variation in the hepatic elimination of drugs an dtoxic chemical agents. In: Annual Review of Chronpharmacology. A Reinberg, M Smolensky and G Labrecque (eds), Vol 4,1-46, Pergamon Press, Oxford, UK. 125. Belanger, P.M., Labrecque, G. 1989. Temporal aspects of drug metabolism. In: Chrono-pharmcology. Cellular and biological interactions. B Lemmer (ed), Marcel Dekker, New York, pp 15-34. 126. Waterhouse, J.M., Minors, D.S. 1989. Temporal asepcts of renal drug elimination. In: Chronopharmacology: Cellular and Biological Interactions, B Lemmer (ed), Marcel Dekker, New York, pp 15-34. 127. Boughattas, N.A., Levi, F., Fournier, C., Lemaigre, G., Roulon, A., Hecquet, B., Mathe, G., Reinberg, A. 1989. Circadian rhythm in toxicities and tissue uptake of 1,2-diammino-cyclohexane (trans-1) oxalatoplatinum (II) in mice. Cancer Res 49,3362-3368. 128. Boughattas, N.A., Levi, F., Hecquet, B., Lemaigre, G., Roulon, A., Fournier, C., Reinberg, A. 1988. Circadian time depencence of murine tolerance for carboplatin. Toxicol Appl Pharmacol 96,233-247. 129. Sinkule, J., Choi, K., Roemeling, R.v., Han, D., Langevin, T. 1986. Preliminary investigations into the circadian variations of doxorubicin (Adriamycin) plasma concentrations and systemic clearance when administered as a prolonged IV infusion. In: Annual Review of Chronopharmacology, A Reinberg, M Smolensky, G Labrecque (eds), Vol 3, Pergamon Press, Ne York, pp 215-216. 130. Aherne, G.W., English, J., Burton, N., Arendt, J., Marks, V. 1987. Chronopharmacokinetics and their relationship to toxicity and effect with reference to methotrexate, 6-mercaptopurine and morphine. Satellite Symposium, Proc Eur Conf Clin Oncol 4,40. 131. Von Mayersbach, H. 1978. Die Zeitstruktur des Organismus. Auswirkungen auf zellul?re Leistungsf?higkeit and Medikamentenemfindlichkeit. Arzneim. Forsch./Drug Res. 28,1824-1836. 132. Burns, R.E. 1981. Circadian rhythmicity in DNA synthesis in untreated mice as a basis for improved chemotherapy. Cancer Res 41,2795-2802. 133. Scheving, L.E. 1984. Chronobiology of cell proliferation in mammals: Implications for basic research and cancer chemotherapy. In: Cell Cycle Clocks, LN Edmunds (ed), Marcel Dekker Publ., New York, NY, pp 455-499 134. Levi, F., Boughattas, N.A., Blazsek, I. 1988. Comparative murine chronotoxicity of anticancer agents and related mechanisms. In: Annual Review of Chronopharmacology. A Reinberg, M Smolensky, G. Labrecque (eds) Vol. 4, Pergamon Press, Oxford, UK, pp 283-332. 135. Walker, P.R., Van Potter, R. 1974. Diurnal rhythms of hepatic enzymes from rats adapted to controlled feeding schedules. In: Chronobiology. LE Scheving, F Halberg, JE Pauly (eds), Igaku Shoin Ltd. Publ, Tokyo, pp 17-22. 136. Von Mayersbach, H. 1980. Rhythms at morphological levels. In: Chronobiology: Principles and Applications to Shifts in Schedules. LE Scheving & F Halberg (eds), Sijthoff and Noordhoff (publ.), Rockville, MD, pp 95-107. 137. Nash, R.E., Echave-Llanos, J.M. 1971. Circadian variations in DNA synthesis of a fast-growing and a slow-growing hepatoma: DNA synthesis rhythm in hepatoma. J. Natl Cancer Inst 47,1007-1012. 138. Burns, R.E., Scheving, L.E., Tsai, T.H. 1979. Circadian rhythms in DNA synthesis and mitosis in normal mice and in mice bearing the Lewis lung carcinoma. Eur J Cancer 15,233-242. 139. Klevecz, R.R., Shymko, R.M., Blumenfeld, D., Braley, P.S. 1987. Circadian gating of S phase in human ovarian cancer. Cancer Res 47,6267-6271. 140. Laerum, O.D., Smaaland, R., Sletvold, O. 1989. Rhythms in blood and bone marrow: potential therapeutic implications. In: Chronopharmacology: Cellular and Biochemical Interactions, B Lemmer (ed), Marcel Dekker, New York, pp 371-393. 141. Buchi, K.N., Moore, J.G., Rubin, N.H. 1987. Circadian cellular proliferation measurements in human rectal mucosa. Chronobiologia 14,155-156. 142. Haus, E., Halberg, F., Scheving, L.E., Cardoso, S., Kuhl, J.F.W., Sothern, R., Shiotsuka, R.N., Hwang, D.S., Pauly, J.E. 1972. Increased tolerance of leukemic mice to arabinolsyl cytosine with schedule adjusted to circadian system. Science 177,80-82. 143. Sothern, R.B., Levi, F., Haus, E., Halberg. F., Hrushesky, W.J.M. 1989. Control of a murine plasmacytoma with doxorubicin-cisplatin: Dependence on circadian time of treatment. J Natl Cancer Int 81,135-145. 144. Hrushesky, W.J.M., Roemeling, R.v., Sothern, R.B. 1989. Circadian chronotherapy: From animal experiments to human cancer chemotherapy. In: Chronopharmacology: Cellular and Biochemical Interactions, B Lemmer (ed), Marcel Dekker, New York, pp 439-473. 145. Mormont, M.C., Boughattas, N., Levi, F. 1989. Mechanisms of circadian rhythms in the toxicity and efficacy of anticancer drugs: Relevance for the development of new analogs. In: Chronopharmacology: Cellular and Biochemical Interactions, B Lemmer (ed), Marcel Dekker, New York, pp 395-437. 147. Halberg, F., Visscher, M.B., Bittner, J.J. 1953. Eosinophil rhythm in mice. Range of occurrance: Effects of illumination, feeding and adrenalectomy. Am J Physiol 174,109-122. 148. Rosenberg, S.A., Grimm, E.A., McGrogan, M., et al. 1984. Biological activity of recombinant human interleukin-2 produced in Escherichia coli. Science 233,1412-1415. 149. Grimm, E.A., Mazumder, A., Zhang, H.Z., et al. 1982. The lymphokine-activated killer cell phenomenon: Lysis of NK resistant fresh solid tumor cells by IL-2 actiated autologous human peripheral blood lymphocytes. J Exp Med 155,1823-1841. 150. Rosenstein, M., Yron, I., Kaufman, Y., et al. 1984. Lymphokine-activated killer cells: Lysis of fresh syngeneic natural killer resistant murine tumor cells by lymphocytes cultured in interleukin-2. Cancer Res 44,1946-1953. 151. Ettinghausen, S.E., Lipford, E.H. III, Mule, J.J., et al. 1985. Recombinant interleukin-2 stimulates in vivo proliferation of adoptively transferred lymphokine-activated killer (LAK) cells. J Immunol 135,3623-3635. 152. Grimm, E.A., Robb, R.J., Roth, J.A., et al. 1983. Lymphokine-activated killer cell phenomena. III. Evidence that IL-2 is sufficient for direct activation of peripheral blood lymphocytes into lymphokine-activated killer cells. J Exp Med 158,1356-1361. 153. Ettinghausen, S.E., Lipford, E.H. III, Mule, J.J., et al. 1985. Systemic administration of recombinant interleukin-2 stimulates in vivo lymphoid cell proliferation in tissues. J Immunol 135,1488-1497. 154. Sondel, P.M., Hank, J.A., Kohler, P.C., et al. 1986. Destruction of autologous human lymphocytes by interleukin-2 activated cytotoxin cells. J Immunol1 37,502-511. 155. Barlozzari, T., Leonhardt, J., Wiltrout, R.H., et al. 1985. Direct evidence for the role of LGL in the inhibition of experimental tumor metastasis. J Immunol 134,2783-2789. 156. Wiltrout, R.H., Herberman, R.B., Zhang, S.R., et al. 1985. Role of organ-associated NK cells in decreased formation of experimental metastases in lung and liver. J Immunol 134,4267-4275. 157. Williams, R., Kraus, L.J., Dubey, D.P., et al. 1980. Circadian bioperiodicity in natural killer cell activity of human blood. Chronobiologia 6,172. 158. Abo, T., Kawate, T., Itoh, K., Kumagai, K. 1981. Studies on the bioperiodicity of the immune response. I. Circadian rhythms of human T, B and K cell traffic in the peripheral blood. J Immunol 126,1360-1363. 159. Levi, F., Canon, C., Blum, J.P., et al. 1985. Circadian and/or circahemidian rhythms in nine lymphocyte-related variables from peripheral blood of healthy subjects. J Immunol 134,217-222. 160. Ritchie, W.S., Oswald, I., Micklen, H.S., et al. 1983. Circadian variation of lymphocyte subpopulations: A study with monoclonal antibodies. Birt Med J 286,1171-1172. 161. Ratajczak, H.V., Sothern, R.B., Hrushesky, W.J.M. 1988. Estrous influence on surgical cure of a mouse breast cancer. J Exp Med 168,88-96. 162. Kern, J.A., Lamb, R.J., Reed, J.C., et al. 1988. Dexamethasone inhibition of interleukin 1 beta production by human monocytes. Post-transcriptional mechanisms. J Clin Invest 81,237-244. 163. Lee, S.W., Tsou, A.P., Chan, H., et al. 1988. Glucocorticoids selectively inhibit the transcription of the interleukin 1-beta gene and decrease the stability of interleukin 1-beta mRNA. Proc Natl Acad Sci 85,1204-1208. 164. Ucker, D. 1988. Cytotoxic T lymphocytes and glucocorticoids activate an endogenous suicide process in target cells. Nature 327,62-67. 165. Sánchez de la Pe?a, S. 1989. Chronomodulation of murine adrenocortical function by interleukin 2. Chronobiologia 16,177. 166. Dinarello, C.A., Wolff, S.M. 1982. Molecular basis of fever in humans. Am J Med 72,799-819. 167. Darzynkiewicz, Z., Williamson, B., Carswell, E., Old, L.J. 1984. Cell cycle specific effects of tumor necrosis factor. Cancer Res 44,83-90. 168. Lotz, M., Vaughan, J.H., Carson, D.A. 1988. Effects of neuropeptides on production of inflammatory cytokines by human monocytes. Science 241,1218-1221. 169. Nakata, K., Kashimoto, S., Yoshida, H., et al. 1988. Augmented antitumor effect of recombinant human interleukin-1-alpha by indomethacin. Cancer Res 48,584-588. 170. Okusawa, S., Gelfand, J.A., Ikejima, T., et al. 1988. Interleukin 1 induces a shock-like state in rabbits. Synergism with tumor necrosis factor and the effect of cyclo-oxygenase inhibition. J Clin Invest 81,1162-1172. 171. Kaushansky, K., Lin, N., Adamson, J. 1988. Interleukin 1 stimulates fibroblasts to synthesize granulocyte-macrophage and granulocyte colony-stimulating factors. Mechanism for the hematopoietic response to inflammation. J Clin Invest 81,92-97. 172. Halberg, F., Johnson, E.A., Brown, B.W., Bittner, J.J. 1960. Susceptibility rhythm to E coli endotoxin and bioassay. Proc Soc Exp Biol Med 103,142-144. 173. Creasey, A.A., Reynolds, M.T., Laird, W. 1986. Cures and partial regression of murine and human tumors by recombinant human tumor necrosis factor. Cancer Res 46,5687-5690. 174. Langevin, T., Young, J., Walker, K., et al. 1987. The toxicity of tumor necrosis factor is reproducibly different at specific times of the day. Proc AACR 28,281. 175. Hrushesky, W., Langevin, T., Nygaard, S., et al. 1987. Circadian stipulation required for reduction of variability in TNF toxicity/efficacy. Intl Conf Tumor Necrosis Factor and Related Cytotoxins. Heidelberg, Sept 14-18. 176. Bocci, V. 1985. Administration of interferon at night may increase its therapeutic index. Cancer Drug Delivery 2,313-318. 177. Abo, T., Kawate, T., Itoh, K., Kumagai, K. 1981. Studies on the bioperiodicity of the immune response. I. Circadian rhythms of human T, B and K cell traffic in the peripheral blood. J Immunol 126,1360-1363. 178. Bertouch, J.V., Roberts-Thomson, P.J., Bradley, J. 1983. Diurnal variation of lymphocyte subsets identified by monoclonal antibodies. Brit Med J 286,1171-1172. 179. Haus, E., Lakatua, D.J., Swoyer, J., Sackett-Lundeen, L. 1983. Chronobiology in hematology and immunology. Am J Anat 168,467-517. 180. Ritchie, A.W.S., Oswald, I., Micklem, H.S., et al. 1983. Circadian variation of lymphocyte subpopulations: A study with monoclonal antibodies. Brit Med J 286,1773-1775. 181. Kawate, T., Abo, T., Hinuma, S., Kumagai, K. 1981. Studies on the bioperiodicity of the immune response II. Covariations of murine T and B cells and a role of corticosteroids. J Immunol 126,1364-1367. 182. Bocci,V. 1985. The physiologic interferon response. Immunology Today 6,7-9. 183. Bocci, V., Paulesu, L., Muscettola, M., Viti, A. 1985. The physiological interferon response. VI. Interferon activity in human plasma after a meal and drinking. Lymphokine Research 4,151-158. 184. Bocci, V. 1985. Distribution Catabolism and Pharmacokinetics of Interferons. In: interferon, NB Finter, R Oldham (eds), Elsevier, AMsterdam, pp 47-72. 185. Blalock, J.E., Smith, E.M. 1980. Human leukocyte interferon: Structural and biological relatedness to adrenocorticotropic hormone and endorphins. Proc Natl Acad Sci 77,5972-5974. 186. Scott, GM. 1983. The toxic effects of interferon in man. In: Interferon, I Gresser (ed), Academic Press, New York, pp 85-114. 187. Scott, G.M., Ward, R.J., Wright, D.J., et al. 1983. Effects of cloned interferon in normal volunteers: Febrile reactions and changes in circulating corticosteroids an trace metals. Antimicr Agents & Chemother 23,589-592. 188. Eskola, J., Frey, H., Molnar, G., Soppi, E. 1976. Biological rhythm of cell-mediated immunity in man. Cl Exper Immun 26,253-257. 189. Cove-Smith, J.R., Kabler, P., Pownall, R., Knapp, M.S. 1978. Circadian variation in an immune response in man. Brit Med J 2,253-254. 190. Abrams, P.F., McClamrock, E., Foon, K.A., et al. 1985. Evening administration of alpha interferon. N Engl J Med 312,443-444, [Letter to the Editor]. 191. Wide, L., Bengtsson, C., Birgegard, G. 1989. Circadian rhythm of erythropoietin in human serum. Br J Haematol 72,85-90. 192. Haus, E., Lakatua, D., Swoyer, J., Sackett-Lundeen, L. 1983. Chronobiology in hematology and immunology. Am J Anatomy 168,467-517. 193. Laerum, O.D., Smaaland, R. 1989. Rhythms in blood and bone marrow: Potential therapeutic implications. In: B Lemmer (ed), Chronopharmacology: Cellular and Biochemical Interactions, Marcel Dekker, New York, pp 371-393. 194. Pasquale, D., Chikkappa, G., et al. 1989. Hydrocortisone promotes survival and proliferation of granulocyte-macrophage progenitors via monocytes/macrophages. Exp Hematol 17,110. 195. Beutler, B., Krochin, N., Milsark, I.W., Luedke, C., Cerami, A. 1986. Control of cachectin (TNF) synthesis: Mechanisms of endotoxin resistance. Science 232,977-980. 196. Gessani, S., McCandless, S., Baglioni, C. 1988. The glucocorticoid dexamethasone inhibits synthesis of interferon by decreasing the level of its mRNA. J Biol Chem 263,7454-7457. 197. Caput, D., Beutler, B., Hartog, K., Thayer, R., Brown-Shimer, S., Cerami, A. 1986. Identification of a common nucleotide sequence in the 3'-untranslated region of mRNA molecules specifying inflammatory mediators. Proc Natl Acad Sci 83,1670-1674. 198. Han, J., Brown, T., Beutler, B. 1990. Endotoxin-responsive sequences control cachectin/tumor necrosis factor biosynthesis at the translational level. J Exp Med 171,465-475. 199. Wood, P.A., Sánchez de la Pe?a, S., Hrushesky, W.J.M. 1990. Circadian dependency of recombinant human erythropoietin (rhEPO) response in the mouse. Proc AACR 31,79 [abst.]. 200. Scheving, L.E., Pauly, J.E., Tsai, T. 1983. The importance of chronobiology in modern research and medicine. Verh Anat Ges 77,59-84. 201. Levi, F., Bailleul, F., Chevelle, C., Benavides, M., Misset, J.L., LeSaunier, F., Dexpas, R., Ribaud, P., Machover, D., Jasmin, C., Regensberg, C., Reinberg, A., Mathe, G. 1987. Chronotherapy of ovarian cancer with 4'-tetrahydropyranyl adriamycin (THP) and cisdichlorodiammine platinum (CDDP). Proc ASCO 6,119. 202. Halberg, F. 1974. Protection by timing treatment according to bodily rhythms--an analogy to protection by scrubbing before surgery. In: Aschoff J, Ceresa F, Halberg F (eds): Chronobiological Aspects of Endocrinology. Milano, Italy: The Publishing House, pp 27-72. 203. von Roemeling, R., Hrushesky, W.J.M. 1987. Circadian shaping of FUDR infusion reduces toxicity even at high-dose intensity. Proc ASCO, 6,293. 204. von Roemeling, R., Hrushesky, W.J.M., Kennedy, B.J., Buchwald, H. 1987. Programmed automatic FUDR chronotherapy improves therapeutic index. Surg Forum 27,401-402.