Supplementary Information (doc 48K)
")
1.
Quantitative reverse transcription PCR based evaluation of ARID1B transcript level.
2μg of total RNA was subjected to reverse transcription in the presence of 2 pico mole ARID1BR primer using Superscript III reverse trancriptase (Invitrogen, Carlsbad, CA, USA) as per manufacterure’s protocol. Appropriate dilutions of the cDNA in triplicate were used for quantitative PCR performed on the SDS 7500 (ABI Inc., Foster city, CA, USA) using the SYBR
Premix Ex Taq (Perfect Real Time kit; Takara Bio Inc., Japan) in presence of ARID1BF and
ARID1BR primers. The PCR conditions were 95
0
C 5min followed by 40 cycles of 94
0
C 30sec;
56 0 C 30 sec; 72 0 C 45 sec. Melting curve analysis was performed to ensure quantitation of specific PCR product. The fold increase in ARID1B transcript level (normalized to GAPDH ) with respect to untreated control was calculated.
2.
Cloning of ARID1B cDNA to generate permanent transfectants in MiaPaCa2 cell line.
Primers bearing Xba I restriction site were used for PCR amplification of KIAA1235 using
Accutaq DNA polymerase (Sigma, USA) as per standard protocol. The PCR product was restricted with Xba I, treated with T4 polynucleotide kinase (NEB, USA) and ligated to Xba I restricted and dephosphorylated (using Shrimp Alkaline Phosphatase, MBI Fermentas, USA) pcDNA3.1HisC (Invitrogen, Carlsbad, CA, USA); the ligation mixture was transformed into
DH5α competent cells. Plasmids isolated from the putative transformants were screened using restriction digestion and confirmed through DNA sequencing.
3.
Various assays performed on the permanent MiaPaCa2 and PANC1 clones.
For crystal violet dye extraction growth assay, 1 × 10
4
cells from each clone were seeded separately in each well of a 12-well plate for 12 hrs, washed twice with 1X PBS and fixed with
20% methanol solution for half an hour at room temperature. Cells were stained with 0.2% crystal violet solution for 30 min at room temperature with gentle shaking. Stained cells were washed with water until a clear background was visible. Crystal violet dye was extracted using
1% SDS and absorbance was determined at 570nm (Varioskan Flash, Thermo Scientific, USA).
MTT cell viability assay was performed as described earlier (Bharadwaj et al., 2008). Briefly,
2,000 cells were seeded and incubated upto 96hr. Absorbance was measured at 590 nm at each time point using a 96-well microplate reader (Bio-Rad, USA). For liquid colony formation assay,
3000 exponentially growing cells (5000 for Panc1 clones) were seeded in 60mm culture dishes
(BD-Falcon, USA) and grown in selection media at 37
0
C. Distinct colonies were observed after two weeks and were subjected to crystal violet staining. Following staining, plates were washed with 1X PBS (until background dye was clear), air dried and images were captured by scanning
(Scanjet 3500C, HP, USA). The visible colonies were counted in Image J software using particle counter. For soft agar assay, 2 ml of bottom agar mix was poured into each well of a 6-well plate and incubated at 37
0
C. On the next day, 10,000 exponentially growing cells for each clone were suspended in 2 ml of Top agar mixture, layered on to the surface of bottom agar and grown for
21 days. A small amount of RPMI media was gently layered on the top agar every alternate day to keep the agar moist. Once the colonies formed, the plates were photographed using a phase contrast inverted microscope (Nikon E2000, Japan), and counted at 4X magnification in ten different fields. Senescence-associated β-galactosidase staining was performed as described
(Debacq-Chainiaux et al ., 2009). Briefly, 1 × 10 4 cells were seeded in a 24-well plate, cultivated
for five days and fixed with 2% formaldehyde/0.2% glutaraldehyde. Cells were stained at 37 °C for 12 h in X-gal staining solution (1 mg/ml X-gal, 40 mM citric acid/sodium phosphate (pH
6.0), 5 mM potassium ferrocyanide, 150 mM NaCl and 2 mM MgCl
2
) and counted under bright field illumination of Evos XL microscope (AMG - Advanced Microscopy Group, USA) in a minimum of three separate fields for each well. Adriamycin (1µM) treated MiaPaCa2 cells were evaluated for comparison.
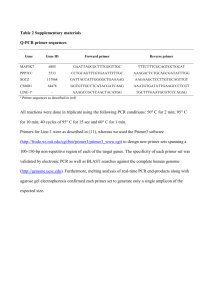
Description of various PCR primers used in this study:
1.
PCR primer pairs for evaluation of ARID1B transcript levels.
Primer Primer Sequence (5’-3’) Ta ( 0 C) 1 Product size (bp)
ARID1BF AGAAAAGTGCCTGGAAGCAG 58 113
ARID1BR ATGCCCAGGTTAGACATTGC
GAPDHF CAATGACCCCTTCATTGACC 58 146
GAPDHR GATCTCGCTCCTGGAAGATG
1
Ta, annealing temperature.
2.
PCR primer sequences for cloning ARID1B cDNA in pcDNA3.1HisC.
Primer Primer Sequence
(5’-3’) 1
Ta ( 0 C) Product size (bp)
Forward TGC TCTAGA CCTTCTCCTGGGGGCCAGAT 60 4458
Reverse TGC TCTAGA TCATAACTGCCCAATCTGAAAC
1
The Xba I restriction site is italicized in both forward and reverse sequence.
3.
PCR primer pairs for evaluation of ARID1B CpG island
Primer Primer Sequence (5’-3’) Ta ( 0 C) Product size (bp)
P1F
P1R
P2F
P2R
P3F
P3R
P4F
TTTTTGTAATTTTTGAGGTTAATAGATAAT 56.5
AACCCAACATAACTTAAAAAACA
TGAAAGTATATAGTAGGAGTAGGTTTAGAG 56.5
AAAAAAAACAATAACCCCCACTT
AAAGTGGGGGTTATTGTTTTTTT
CCTCCAAATCCCGAACTC
TGAATAGTGTGTATTATTATTTTTTGTTTT
56.3
53.9
337
156
182
256
P4R CAATTTATAATTAAAAAACAACCCC
References:
Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. (2009). Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo . Nat Protoc 4: 1798–1806.
Bharadwaj U, Li M, Chen C, Yao Q. Mesothelin-induced pancreatic cancer cell proliferation involves alteration of cyclin E via activation of signal transducer and activator of transcription protein 3. Mol Cancer Res. 2008;6:1755-1765.