Comparing Sequence and Structure-based Classifiers for Predicting
advertisement

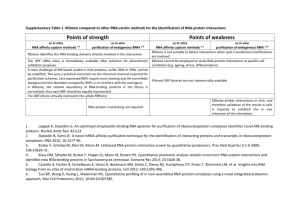
Title: Comparing Sequence and Structure-based Classifiers for Predicting RNA Binding Sites in Specific Families of RNA Binding Proteins Authors: Michael Terribilini1, Cornelia Caragea2, Deepak Reyon3, Jeffry Sander4, Jae-Hyung Lee5, Robert L. Jernigan6, Vasant Honavar7, and Drena Dobbs8 Short Abstract We evaluate machine learning classifiers for predicting RNA-binding residues in proteins, using either sequence-based information only, or a combination of sequence and structure-derived information and quantitate relative contributions of these different input types to overall prediction performance. We also present novel classifiers optimized for specific families of RNA binding proteins. Abstract Protein-RNA interactions play critical roles in a wide range of biological processes. Previously, we developed a machine learning approach for predicting which amino acids of an RNA-binding protein mediate protein-RNA interactions, using only the amino acid sequence of the protein as input (Terribilini et al, 2006, RNA 12:1450; http://bindr.gdcb.iastate.edu/RNABindR/). Here we report an evaluation of the relative contributions of sequence, structural features and evolutionary information to performance of algorithms for predicting RNA binding residues in proteins. In this study we train and test multiple classifiers using several benchmark datasets, including a non-redundant dataset of 181 RNA-binding polypeptide chains with <30% sequence identity (RB181), and “custom” datasets comprising sets of related RNA-binding proteins. We systematically compare results obtained using simple classifiers that use only one type of information as input (e.g., Naïve Bayes classifier, using only amino acid sequence as input) with results obtained using ensemble classifiers that exploit specific combinations of input information (e.g., an ensemble of Naïve Bayes classifiers that use the amino acid sequence, information from sequence homologs and/or the identities of spatial neighbors in known structures as input). Our results, partially summarized in Table 1 below, indicate that the best “overall” performance, evaluated on the basis of AUC for ROC curves, is obtained using ensemble classifiers using amino acid sequence information in combination with either: i) PSSMs (derived from sequence homologs identified using BLAST) or ii) spatial neighbor information (extracted from PDB structures of proteins). Also, we will report results obtained using “custom” classifiers for predicting RNA-binding residues in specific families of RNA binding proteins (i.e., those sharing similar sequences or structures). Table 1: Comparison of Classifiers for RNA-binding Site Prediction Sequencebased Area Under ROC Curve 1 2 3 4 5 6 7 8 0.74 Structurebased 0.77 PSSM-based 0.80 Bioinformatics & Computational Biology, Iowa State University, E-mail: terrible@iastate.edu Computer Science, Iowa State University, E-mail: caragea@cs.iastate.edu Bioinformatics & Computational Biology, Iowa State University, E-mail: dreyon@iastate.edu Bioinformatics & Computational Biology, Iowa State University, E-mail: jdsander@iastate.edu Bioinformatics & Computational Biology, Iowa State University, E-mail: jhlee777@iastate.edu Biochemistry, Biophysics & Molecular Biology, Iowa State University, E-mail: jernigan@iastate.edu Computer Science, Iowa State University, E-mail: honavar@cs.iastate.edu Genetics, Development, & Cell Biology, Iowa State University, E-mail: ddobbs@iastate.edu Ensemble 0.81 Scientific Justification 1. The ability to reliably predict which residues of a protein directly contribute to RNA binding would significantly enhance our understanding of how proteins recognize RNA and potentially generate new strategies for clinical intervention in both genetic and infectious diseases. We will present our results on predictions made on several clinically important RNA-binding proteins, such as the HIV-1 Rev and human telomerase reverse transcriptase (hTERT) protein. We will also present a comparison of our results with those of other labs 2. Classifiers can be "tuned" to enhance either specificity or sensitivity of interface residue prediction for specific subfamilies of related RNA binding proteins, thus facilitating the design of experimental investigations.