Introduction to Systematics - BioQUEST Curriculum Consortium
advertisement

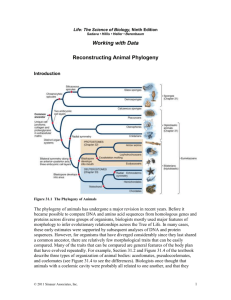
Introduction to Systematics Fall 2007 The following introduction is slightly modified from Brown et al. 2003. Tree of Life: Microbial Evolution in Microbes Count! John R. Jungck, Ethel D. Stanley, and Marion Field Fass, Eds. Published by BioQUEST Curriculum Consortium and American Society for Microbiology. For the next few weeks we’ll be looking at all the organisms that are capable of photosynthesis. Our job is to determine what evolutionary pattern led to their diversification. There are two types of data we can use. The first is morphological and the second is DNA. Ideally these two methods should give us similar results. Today we’ll be acquainting ourselves with two software packages: Biology Workbench and PHYLP. DNA and phylogenetic trees: Background Bioinfomatics is a relatively new field that involves the use of computers to mine the incredible amount of molecular information available to scientists. This information includes DNA and protein sequences as well as other types of information for a wide variety of organisms. Phylogeny reconstruction is an important application of bioinformatics. Phylogeny refers to a hypothesis for the evolutionary history of a group of species. Phylogenetic trees are usually constructed from sequence data but may also be based on morphological characteristics, and metabolic characteristics, or other forms of molecular data (e.g. DNA fingerprint profiles). Phylogenies are typically represented as resolved or partially resolved bifurcating trees, consisting of nodes and edges (Fig. 1a, 1b). The relative lengths of different edges represent the degrees of relatedness, and are often shown in proportion (weighted tree, as in Fig. 1a). However, if the investigator is only interested in the respective groupings of species sometimes the edges are shown as being of an equal arbitrary length (unweighted tree, as in Fig. 1b). Fig. 1a. An unrooted weighted tree. Fig. 1b. An unrooted unweighted tree. Evolutionary tree building seeks to find the tree that makes the most biologically-sensible explanation of how a mixed group of species evolved. There can be many possible bifurcating tree shapes to consider for even relatively small numbers of species compared. Some methods of tree building use some secondary criteria to evaluate how well each of the possible tree shapes fit the data. However, an alternative approach, which works better for larger numbers of species is to first construct a table containing measures of difference between pairwise comparisons of species (e.g. perhaps count the number of mutations that differ between the sequences of different species). The next step then uses the values in the table to determine the order species are clustered into a bifurcating tree (Fig. 2). 1 8 7 1 2 4 5 2 8 6 3 4 5 1 7 8 6 3 1 7 2 2 7 8 6 5 3 6 3 4 Fig. 2 Phylogenetic tree building by pairwise comparisons. This procedure produces just one tree, and does not evaluate this tree against other possible fully resolved bifurcating trees, which may be almost as equally probable. There are several factors to consider when constructing a tree: 5 Did the data evolve on a bifurcating tree? Can we expect the patterns in the data to indicate only 1 unique tree? How far back in history can we reliably infer phylogeny from sequence data? Can we infer a species’ phylogeny from the comparative analysis of only small sections of the genomes of our organisms? Will protein or DNA or rRNA sequences be better for inferring phylogeny? Has the data evolved under a molecular clock? Is it valid to compare homologous sequences if they are from different genome compartments (e.g. from chloroplasts, mitochondria, nuclei, nucleomorphs)? How should we think about sequence evolution - what are the properties of sequence evolution? Different models of sequence evolution can be used to correct the difference values in our starting distance table. Different corrections could result in different values that change the shape of our final tree. What are the assumptions of these models? Which ones would be best? How would I decide? Bioinfomatics Tools The purpose of this exercise is to develop a basic understanding of bioinfomatics and learn some of the skills involved in constructing and interpreting phylogenetic trees and cladograms. The basic tools consist of a bank of data, in our case DNA sequences, and a program to compare the similarities and differences between sequences. These programs also display the results in the form of a tree. Simply generating the tree is not the end of the story. Interpretation is a time-consuming and extensive process and you will need to spend a fair amount of time interpreting your trees. Learning the basic skills Follow your instructor’s directions and complete the “warm-up” activity. 1. Your characteristic grid for your “organisms”: Organism 1 2 3 4 5 6 Char 1 Char 2 Char 3 Char 4 Char 5 Character name 1______________________ Codes :____________________________ Character name 2______________________ Codes :____________________________ Character name 3______________________ Codes :____________________________ Character name 4______________________ Codes :____________________________ Character name 5______________________ Codes :____________________________ Your phylogeny based on morphological characteristics: Now weight your characteristics as ancestral or derived. What does your phylogeny look like now? What changes did you make? For the next part of the lab you will work with the 10 species you selected last week at Lamberton Conservatory. First complete the following chart using the information you collected last week. Your characteristic grid for the Lamberton species: Organism Char 1 Char 2 Char 3 Char 4 Char 5 1 2 3 4 5 6 7 8 9 10 Character name 1______________________ Codes :____________________________ Character name 2______________________ Codes :____________________________ Character name 3______________________ Codes :____________________________ Character name 4______________________ Codes :____________________________ Character name 5______________________ Codes :____________________________ Using your phylogeny from last week, and information on phylogenies you have learned in this lab, draw your phylogeny based on morphological characteristics (state whether you are using pure morphological data, or are designating some characteristics ancestral or derived. Step by step instruction 1. Get on to the internet and type in http://workbench.sdsc.edu/ 2. Click on “Set up a free account” and follow the instruction to set up an account. Be sure to keep a record of your name and password. Next time you enter, click on “Enter the Biology Workbench 3.2” and all you have to do is to type your name and password. 3. Once you are in, click on “Nucleic Tools”. 4. Highlight “Ndjinn – Multiple Database Search”. Click “Run”. 5. There many databases available. For our project we will use GBPLN (GenBank Plant Sequences, which includes the fungi and algae.) For this week, we will be working on the10 plants you chose at the Lamberton Conservatory last week in lab. 6. Click on “GBPLN”. (You have to scroll down for this option.) 7. Scroll up and type in the scientific name for the first species on your list. Click on “Show 10 hits”. 8. Click on “Search”. You will see a list of choices. Scroll down and make notes on what types of sequences are available. What genes are sequenced? Are they whole or partial sequences? Use the grid on the next page to help you organize these data. Enter the gene name and whether it is a partial sequence of a complete sequence. Organism Gene Gene Gene Gene Gene 1 2 3 4 5 6 7 8 9 10 9. Repeat this process for all your species, then determine what gene sequence is available for most of your species and import those sequences to your account. 10. Import sequences by searching again and highlighting the line for the data you want. Click on the “Import Sequence(s)” button located at the end of the first line of the interactive box. 11. Now we are ready to generate a tree. Select all the organisms. Click “Run”. All the boxes in front of these organism names should be checked. 12. Using the scroll box and scroll down and highlight “CLUSTALW – Multiple Sequence Alignment”. Click “Run”. 13. A new screen will appear. You can choose to make a rooted or unrooted tree by clicking on the arrow next to the box labeled “Guide tree display” and choosing rooted or unrooted. Then, click “Submit”. The screen will go blank and you may have to wait several minutes. Wait until a screen titled “CLUSTALW” with “Sequence alignment” appears. Scroll down to examine the DNA sequences and how they align with each other. Scroll further down and you will see your tree! 14. Open Microsoft Word. Click “Edit”, then “Paste” and your tree will reappear. Adjust the size of your tree by selecting the tree image and resize from the lower right corner so two trees can fit on a page. Type in a label for each tree using consecutive figure numbers. 15. Look up each GBPLN number and write the corresponding species beside the number by hand. You may also label each one by pulling down the “Insert” menu then selecting and releasing on “TextBox”. Click where you want the textbox to be located and type the name of the species. Drag the textbox next to the corresponding number. How does this tree compare with the one you drew last week and the phylogeny you drew earlier in this lab, based on morphological characteristics? Morphological and molecular data, and phylogenetic trees. To complete a phylogenetic tree using character states requires a lot of homework. In the weeks to come, we’ll be building a character matrix with the following headings: Taxon Characters 1 2 3 4 5 6 7 8 9 10 11 Phylum1 Phylum2 Phylum3 Phylum4 For each character you need to name the character and determine how many different states it will have, then assign a number to each of the character states. Those are the numbers you will enter in your character matrix. Today you’ve had some practice. Starting next week, we’ll start building the phylogeny of all photosynthetic organisms. Working in teams of four, use your text book and list all the phyla that your text discusses. Look at the pictures and draw on your previous knowledge of plants. What are some characteristics that you notice many, or some phyla have? For example, we learned that having vascular tissue was a major evolutionary advantage. So, one of our characters could be “vascular tissue” with two character states, 0=absent and 1=present. See what other characters you want to include. We’ll refine these lists as we proceed in the semester. Build a character matrix list in Excel with the following columns: character name, character description, character state 0, character state 1, etc. for all character states for that character, and why it’s an important character for determining evolutionary relationships in plants. Fill a row in for each character. Here’s a sample for the example given above: Character name vascular tissue Character description vascular tissue includes presence of xylem and phloem Character state descriptions 0 = no vascular tissue 1 = vascular tissue present Why is this important? vascular tissue allowed plants to grow larger and become more complex… Next character listed here For next week turn in: Your Lamberton list with a photograph of each species (properly cited) This completed lab handout Your tree from Biology Workbench. A printed copy of your proposed character matrix for all photosynthetic life with at least 5 characters described as above. rev. 6/19/08 bjb