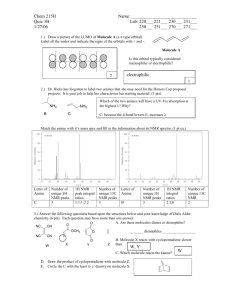
Synthesis, Determination, and Catalytic Measurement of Ruthenium Indenylidene
Complexes used in Olefin Metathesis
Author: Adam Capriola
CHM 2521 Section 151, Department of Chemistry, Saint Joseph’s University
Date Submitted: April 19, 2010
Abstract
The reaction of RuCl2(PPh3)2, THF, and diphenylpropargyl alcohol under reflux yields
C51H40P2Cl2Ru in 46% yield. 1H NMR spectroscopy of C51H40P2Cl2Ru shows a series of
overlapping peaks at δ 7.3-7.8. C51H40P2Cl2Ru can then react with dichloromethane and
tricyclohexylphosphine to form C51H76Cl2P2Ru. 1H NMR spectroscopy of C51H76Cl2P2Ru yields
the same series of peaks found around δ 7.3-7.8 that C51H40P2Cl2Ru exhibits, along with a faint
series of peaks at δ 1.8-2.1.
31
P NMR spectroscopy of both products shows a single peak around
δ 29.5. This suggests what was believed to be C51H76Cl2P2Ru was actually mostly
C51H40P2Cl2Ru. Catalytic measures of the two synthesized products were inconclusive due to
their similar natures, however, it is expected that C51H76Cl2P2Ru is the better catalyst as it has
bulkier, more readily dissociating substituents.
Introduction
The reaction of RuCl2(PPh3)2 with THF and diphenylpropargyl alcohol under reflux
yields C51H40P2Cl2Ru.1 The reaction specifically takes place in the following manner:
Scheme 1
OH
Ph
Ph
PPh3
Ru PPh3
Cl
PPh3
Cl
THF, Reflux
PPh3
Cl
Ru
Cl
PPh3
Ph
C51H40P2Cl2Ru can then react with dichloromethane and tricyclohexylphosphine to form
C51H76Cl2P2Ru. The reaction occurs in the following manner:
Scheme 2
PPh3
Cl
Ru
Cl
PPh3
Ph
PCy3
CH2Cl2
PCy3
Cl
Ru
Cl
PCy3
Ph
These products be distinguished via 1H and 31 NMR spectroscopy. The 1H NMR spectrum of the
C51H76Cl2P2Ru will yield peaks representative of the newly added cyclo groups, which are
missing in C51H40P2Cl2Ru. The 31P NMR spectra of each product should theoretically each show
1 peak, with the peak of C51H76Cl2P2Ru being downfield from C51H40P2Cl2Ru because of the
lower electron density around the phosphorus.
The products from these two reaction are of interest because they are ruthenium alkidene
complexes, which are alternatives to Grubbs’ catalysts and are much less difficult to prepare in
the laboratory.1 These two ruthenium indenylidene complexes can be used as catalysts in ring
closing metathesis. Show below are the balanced reaction and mechanism in which diethyl
diallylmalonate undergoes this process with the aid of a ruthenium catalyst:
Scheme 3
CO2Et
EtO2C
Ru Catalyst
EtO2C
CO2Et
+
H2C CH2
CH2Cl2
Scheme 4
PCy3
Cl
Ru
Cl
PCy3
Ph
EtO2C
CO2Et
EtO2C
+
PCy3
Cl
Ru
Cl
PCy3
Ph
EtO2C
+PCy3
EtO2C
PCy3
Cl
Ru
Cl
Ph
EtO2C
+
EtO2C
PCy3
Cl
Ru
Cl
Ph
EtO2C
EtO2C
Ph
-PCy3
Cl Cl
Ru PCy
EtO2C
3
Cl Cl
Ru PCy
3
EtO2C
EtO2C
PCy3
Cl
Ru
Cl
PCy3
+PCy3
-PCy3
PCy3
Ru
Cl
Cl
+
EtO2C
CO2Et
The relative catalytic rates of the two ruthenium indenylidene complexes can be monitored via
GC/MS. Determination of the starting material and product from this technique can show the
relative percentages of each material within a solution. By comparing the ratio of reagent to
product for each of the ruthenium complexes, it can be determined which is a better catalyst, as
the more efficient catalyst will sport the lower ratio of reagent to product.
Experimental
All syntheses were carried out in nitrogen and the reagents and solvents were purchased
from commercial sources and used as received unless otherwise noted. The synthesis of
C51H40P2Cl2Ru (1A), C51H76Cl2P2Ru (1B), and C11H16O4 (2) were based on reports published
previously.1
C51H40P2Cl2Ru (1A). A hot, dry 100 mL 3 neck round bottom flask was obtained from
an oven and connected to it were a cold water condenser, septum, and sidearm stopcock. A gas
inlet was connected to the condenser and a bubbler was connected to the gas inlet. All joints
were greased. A stir bar was placed in the round bottom flask and the apparatus was connected
to a nitrogen source. The condenser was connected to a cold water source. The round bottom
flask was degassed with N2 until cool, at which time RuCl2(PPh3)2 (0.179 g, 1.87 x 10-4 mol),
THF (10 mL), and diphenylpropargyl alcohol (0.080 g, 3.84 x 10-4 mol) were subsequently
added to the reaction vessel. A sand bath was constructed and was used to heat the solution.
The sand bath was set to 80% power and the mixture began to reflux a while later, but THF
began to evaporate over time so the sand bath was turned down to around 40% power and an
additional 30 mL of THF had to be added to the solution during the 2.5 h reflux period. The stir
bar was spun at a moderate speed during this time.
After the reflux period had been completed, the reaction mixture was allowed to cool to
room temperature. The solution was then transferred to a single neck 50 mL round bottom flask
at which time the solution was taken off the N2 supply and was exposed to air for the remainder
of the synthesis. The solvent was removed via rotary evaporation leaving a thick, dark, brownish,
reddish liquor. 1.5 mL dichloromethane was added to the liquor along with 9 mL hexane, which
was slowly pipeted in. A dark red solid was precipitated and filtered using a small fritted funnel
and was washed 3 times with about 2 mL hexane during each rinsing. The solid was vacuum
dried and placed into a pre-weighed vial (9.698 g). The vial was stored in a dessicator for 1
week. The final weight of the vial was 9.767 g. The product was determined to be 1A (0.069g,
7.78 x 10-5 mol, 41.6% yield based on the amount of RuCl2(PPh3)2 used). 1H NMR (CDCl3): δ
7.3-7.8 (6 H, overlapping signal, Ph and indenylidene).
31
P NMR (CDCl3): δ 29.5 (s, Ru-P).
FTIR (ATR) ν 1928 cm-1 (m).
C51H76Cl2P2Ru (1B). A hot, dry 100 mL 3 neck round bottom flask was obtained from
an oven and connected to it were a cold water condenser, septum, and sidearm stopcock. A gas
inlet was connected to the condenser and a bubbler was connected to the gas inlet. A stir bar was
placed in the round bottom flask and the apparatus was connected to a nitrogen source. The
condenser was connected to a cold water source. The round bottom flask was degassed with N2
until cool, at which time 1A (0.050 g, 5.64 x 10-5 mol), dichloromethane (7 mL), and
tricyclohexylphosphine (0.055 g, 1.96 x 10-4 mol) were subsequently added to the reaction vessel.
The mixture was stirred at a moderate speed at room temperature for 1.5 h. 2 mL of additional
dichloromethane was added to the solution during this time as some had evaporated off.
The solution was then transferred to a 50 mL single neck round bottom flask at which
time the solution was taken off the N2 supply and was exposed to air for the remainder of the
synthesis. The solvent was removed via rotary evaporation. The remaining solid was suspended
with 5 mL of hexane. This new solution was stirred at a moderate speed at ambient temperature
for 0.5 h. The resulting solid was filtered using a small fritted funnel and was washed 3 times
with about 2 mL hexane during each rinsing. The solid was vacuum dried and placed into a preweighed vial (9.737 g). This vial was stored in a dessicator for 1 week. The final weight of the
vial was 9.824 g. The product was determined to be 1B (0.087g, 9.42 x 10-5 mol, 167% yield
based on the amount of 1A used). 1H NMR (CDCl3): δ 1.8-2.1 (5 H, overlapping signal, PCy3),
δ 7.1-7.9 (6 H, overlapping signal, Ph and indenylidene).
31
P NMR (CDCl3): δ 29.8 (s, Ru-P).
FTIR (ATR) ν 1921 cm-1 (m).
C11H16O4 (2). A hot, dry 100 mL 3 neck round bottom flask was obtained from an oven
and connected to it were a cold water condenser, septum, and sidearm stopcock. A gas inlet was
connected to the condenser and a bubbler was connected to the gas inlet. A stir bar was placed in
the round bottom flask and the apparatus was connected to a nitrogen source. The condenser
was connected to a cold water source. The round bottom flask was degassed with N2 until cool,
at which time 1B (0.010 g, 1.08 x 10-5 mol), anhydrous dichloromethane (6 mL), and diethyl
diallylmalonate (0.100 g, 4.16 x 10-4 mol) were subsequently added to the reaction vessel. The
mixture was stirred at a moderate speed at room temperature for just over 1 h. The solution was
then transferred to a 25 mL single neck round bottom flask at which time the solution was taken
off the N2 supply and was exposed to air for the remainder of the synthesis. The solvent was
removed via rotary evaporation. 1H NMR (CDCl3): δ 1.2 (t, -CH3), δ 2.6 (d, -CH2), δ 4.1 (q, OCH2), δ 5.1 (m, =CH2), δ 5.6 (tt, C-H), δ 6.8-7.7 (6 H, overlapping signal, Ph and indenylidene).
GC-MS (CH2Cl2): 212 (2.5%, (2)), 241 (82.2%, (3)).
The process described above was repeated by a laboratory partner using 1A in lieu of 1B.
1
H NMR (CDCl3): δ 0.9 (t), δ 1.25 (s), δ 1.55 (s), δ 1.84 (t), δ 3.74 (t), δ 6.8-7.7 (6 H,
overlapping signal, Ph and indenylidene). GC-MS (CH2Cl2): 212 (0.12%, (2)), 241 (59.1%, (3)).
C13H20O4 (3). The 1H NMR spectrum of (3) was obtained from Sigma Aldrich.2 1H
NMR (CHCl3): δ 1.25 (t, -CH3), δ 2.6 (d, -CH2), δ 4.2 (q, O-CH2), δ 5.1 (m, =CH2), δ 5.7 (tt, CH).
Results
The reaction of RuCl2(PPh3)2 with diphenylpropargyl alcohol yielded 0.069g of product,
which was determined to be 1A. This translated to 7.78 x 10-5 mol and thus a 41.6% yield based
on the amount of RuCl2(PPh3)2 used, which was the limiting reagent in the reaction. Proton
NMR spectroscopy of 1A yielded one series of peaks of interest. From δ 7.3-7.8 there was a
sequence of peaks representing the 6 different aromatic hydrogens from the phenyl and
indenylidene groups.
31
P NMR spectroscopy elicited one peak at δ 29.5 which can be attributed
to phosphorus coordinated with the metal, Ru. The IR spectrum of the substance gave one
notable peak at 1928 cm-1, but the identity of this peak was unable to be determined.
The reaction of 1A with dichloromethane and tricyclohexylphosphine yielded 0.087g of
product, which was determined to be 1B. This translated to 9.42 x 10-5 mol and thus a 167%
yield based on the amount of 1A used, which was the limiting reagent in the reaction. Proton
NMR spectroscopy of 1B yielded two series of peaks of interest. From δ 1.8-2.1 were noted a
faint sequence of overlapping signals, which were thought to be due to the 5 different hydrogens
from the PCy3 groups. From δ 7.1-7.9 there was a string of peaks representing the 6 different
aromatic hydrogens from the phenyl and indenylidene groups.
31
P NMR spectroscopy elicited
one peak at δ 29.8 which can be attributed to phosphorus coordinated with the metal, Ru. The IR
spectrum of the substance gave one notable peak at 1921 cm-1, but again the identity of this peak
was unable to be determined.
The reaction using 1B as a catalyst to perform ring closing metathesis on diethyl
diallylmalonate produced a product with a 1H NMR spectrum containing several peaks of
interest. The triplet δ 1.2 was thought to be due to the methyl group, the doublet at δ 2.6 was
thought to be due to –CH2 groups, the quartet at δ 4.1 was thought to be due to the O-CH2 groups,
the multiple peaks at δ 5.1 were thought to be from =CH2, the triplet of triplets at δ 5.6 was
thought to be from C-H, and lastly the extremely weak overlapping signals at δ 6.8-7.7 were
thought to be from phenyl and indenylidene groups. These assumptions are made taking into
consideration that the 1H NMR spectrum of diethyl diallylmalonate was identical, save for the
almost negligible peaks from δ 6.8-7.7.2 The GC/MS of 1B gave what were thought to be
signals of interest at times 6.648 min and 6.945 min. The reading at 6.648 min accounted for
2.5% of the scan and was thought to be C11H16O4 because its m/z of 212 appeared as a peak. The
reading at 6.945 min accounted for 82.2% of the scan and was thought to be diethyl
diallylmalonate because its m/z of 241 appeared as a peak, albeit very small. This gave a
proposed ratio of 33:1, reactant to product.
When using 1A as the catalyst in lieu of 1B in this reaction, proton NMR spectroscopy of
the product elicited several peaks, most of which were not able to be identified. The sequence of
overlapping peaks from δ 6.8-7.7 was attributed to the 6 different hydrogens from phenyl and
indenylidene groups, but the triplet at δ 0.9, the singlet at δ 1.25, the singlet at δ 1.55, the triplet
at δ 1.84, and the triplet at δ 3.74 could not be determined. A standard 1H NMR spectrum of the
desired product C11H16O4 was unobtainable for comparison. The GC/MS of 1A gave what were
thought to be signals of interest at times 6.648 min and 6.974 min. The reading at 6.648 min
accounted for 0.12% of the scan and was thought to be C11H16O4 because its m/z of 212 appeared
as a peak. The reading at 6.974 min accounted for 59% of the scan and was thought to be diethyl
diallylmalonate because its m/z of 241 appeared as a peak, again albeit very small. This gave a
proposed ratio of 491:1, reactant to product.
Discussion
The results of this experiment are inconclusive. The first reaction seemed to give a
decent percent yield of 1A and it was identifiable through 1H and 31P NMR spectroscopy,
however there were a few erroneous peaks noted on the 1H NMR spectrum and the peaks of
interest were somewhat weak. The 31P NMR spectrum of 1A was inconclusive at first, so a new
scan was done at a later time with a different sample. These facts seem to suggest that the
original 1A obtained was not very pure. During the procedure, the sand bath was not adequately
controlled, and this is most likely what caused the impure product. Because the reaction was
overheated, side products may have formed or the original reagents did not react to completion,
and in turn, the percent yield was in reality not as high as it appeared. This also attributes to the
extra peaks that showed up on the 1H NMR spectrum. The oxidation state of 1A is +4 and its
electron count is 16.
The second reaction resulted in a percent yield of 167% of what was thought to be 1B,
which again suggests some error. The 31P of this product gave a peak in nearly the exact same
position as 1A, so this seems to confirm that the product obtained from the second reaction was
not 1B, but mostly 1A. The peak should have shifted downfield to about δ 41, which is what
colleagues have reported. The proton NMR spectrum does show faint peaks from δ 1.8-2.1
which is where one would expect hydrogens attached to non-aromatic cyclo groups to be found.
This means that some of the -PPh3 groups did convert to -PCy3 groups, but a significant amount
on the whole. The IR spectra of the products after reactions one and two are also quite similar,
again hinting that nothing really transpired during reaction two. The procedure during reaction
two went as detailed by the laboratory manual, so this means the starting material was probably
impure and thus could not react to completion.1 The oxidation state of 1B would also be +4 with
an electron count of 16.
Because reaction one and reaction two seemed to yield the same product, the ring closing
reactions cannot accurately be compared for catalytic activity. Theoretically 1B is the better
catalyst, as it has PCy3 ligands opposed to the PPh3 ligands characteristic of 1A. PPh3 ligands
have a cone angle of 145o while PCy3 ligands have a cone angle of 170o.3 The larger the cone
angle, the bulkier the ligand and the faster it dissociates, allowing for expedited ring closing
metathesis.3 The mechanism in which this takes place can be seen in Scheme 4. The reaction of
diethyl diallylmalonate with 1B did not seem to elicit the ring closing mechanism. The 1H NMR
spectra of the product looks identical to that of the starting material, diethyl diallylmalonate, save
for one area around δ 6.8-7.7 where traces of what looks like aromatic structures, namely phenyl
and indenylidene groups can be found. It looks like there was such a minute amount of catalyst
available that it never interacted with diethyl diallylmalonate to close the ring.
GC/MS of 1B shows two signals which may account for diethyl diallylmalonate and the
closed ring. At time 6.648, a peak accounting for 2.5% all that was picked up by the scan
contains a signal of 212 can be seen which corresponds with the m/z of C11H16O4. At time 6.945
min, a peak accounting for 82.2% of the scan contains a signal of 241 can be seen which
corresponds with the m/z of diethyl diallylmalonate. This gives a ratio of 33:1, reactant to
product, which means the yield was rather poor. It does seem to suggest that some product may
have been formed, however product was not visible on the 1H NMR spectrum, so this
interpretation may be inaccurate.
The reaction of diethyl diallylmalonte with 1A yielded a different 1H NMR than the
reaction with 1B did. It also shows overlapping peaks at δ 6.8-7.7 indicative of phenyl and
indenylidene groups, but these peaks are much more noteworthy, meaning there was an
abundance of catalyst available, where in the other reaction there was almost no catalyst
available. Hence, upfield peaks are seen and are believed to be product, but these peaks are
unable to be confirmed. A standard 1H NMR spectrum of C11H16O4 is unobtainable for
comparison. The peaks reminiscent of the starting material seen in the 1H NMR spectrum for the
reaction with 1B are not visible, which means there was some sort of change in the starting
material.
GC/MS analysis however does not seem to confirm the presence of a closed ring product.
At time 6.648, a peak accounting for 0.12% all that was picked up by the scan contains a signal
of 212 can be seen which corresponds with the m/z of C11H16O4. At time 6.974 min, a peak
accounting for 59% of the scan contains a signal of 241 can be seen which corresponds with the
m/z of diethyl diallylmalonate. This gives a ratio of 491:1, reactant to product, which means the
almost no product formed at all despite the presence of what seems to be a copious amount of
catalyst. The reaction with 1B has a ratio of 33:1 and had almost no visible catalyst in its 1H
spectrum, so it could be possible that the GC/MS was analyzed improperly.
The sources of error are difficult to pinpoint, but one issue may have been the flow of
nitrogen through the system. If the flow was too great, solvent would have been lost and it
would have hindered the reactions. If the hot 100 mL three neck round bottom flask was not
allowed to cool completely, that may have also caused a side reaction to occur due to the
unwarranted heat. Also, as noted earlier during the synthesis of 1A, the reaction was overheated,
which could have caused side products to form and thus inhibit the results of the following
syntheses.
Conclusion
The main purposes of the experiments were to synthesize 1A and 1B, confirm their
structures via 1H, 31P, and IR spectroscopy, and to determine their relative catalytic rates during
ring closing metathesis of diethyl diallylmalonate. 1A was identifiable by a series of overlapping
peaks at δ 7.3-7.8 representative six different hydrogens attached to phenyl and indenylidene
groups. This material was collected in a 46% yield, but in reality the yield was likely lower due
to contaminants. 1B was synthesized with 167% yield, which suggests error. It was vaguely
identifiable through its 1H NMR spectrum by a series of peaks found at δ 1.8-2.1 representative
of protons attached to cyclo groups, namely the PCy3 substituents. This spectrum also contained
the same series of overlapping peaks found around δ 7.3-7.8 for 1A. The 31P NMR and IR
spectra for 1A and 1B are nearly identical, suggesting that almost no change in structure took
place during the synthesis of 1B from 1A.
Because 1B did not properly synthesize, or did in an extremely low proportion, it was not
feasible to measure 1A and 1B in comparison of their catalytic properties. It would be expected
that 1B would be a better catalyst, as it contains bulkier groups which in theory dissociate faster.3
The 1A and 1B synthesized were both used as catalysts for ring closing metathesis of diethyl
diallylmalonate. The product from the synthesis with 1B gave a 1H spectrum nearly identical to
that of the starting material, diethyl diallylmalonate, which says that there was too low of a
concentration of catalyst for the reaction to occur in the time allotted. The product from the
synthesis with 1A gave a different 1H with peaks that are thought to be the desired product, but
no standard 1H NMR spectrum of the product is obtainable. The results from the GC/MS of both
products runs contrary to the belief that any significant amount of C11H16O4 was synthesized at
all, and thus the results from this laboratory experiment are inconclusive.
References
(1) Pappenfus et al. Synthesis and Catalytic Performance of Ruthenium Carbene Complexes for
Olefin Metathesis: A Microscale Organometallic Experiment. Journal of Chemical Education.
2007, 84, 1998-2000.
(2) http://www.sigmaaldrich.com/spectra/fnmr/FNMR005436.PDF
(3) Miessler, G. L.; Tarr, D. A. Inorganic Chemistry: Third Edition. Pearson Prentice Hall:
Upper Saddle River, 2004; pp 523-546.