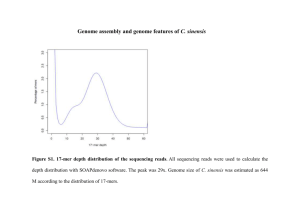
Detail of the upgraded genome
advertisement

Detail of the upgraded genome Genomic features of C. sinensis Besides paired-end data previously published [1], we sequenced two paired-end and two mate-pair libraries again. In total, 263.38 million raw pairs of sequence reads (107X coverage) were produced,with 221.62 million (63X coverage) used for genome assembly after data filtering (Table S1). The assembled genome includes 4,348 scaffolds with a total length of 550 Mb. The upgraded version, with an N50 scaffold/contig length of more than 417/233 kb and the largest scaffold of more than 2 Mb, was improved compared with the previously published version (Table 1). The GC content was calculated to be approximately 43% (Figure S4). In C. sinensis, approximately 32% of the genome represents interspersed repeats, based on both known and ab initio repeat libraries (Figure S5). To estimate the gene coverage of the assembly, CEGMA core genes [2] and assembled transcript data were mapped to the C. sinensis genome. As a result, approximately 97% of the transcripts were mapped, and on average, approximately 83% of the assembled transcripts were mapped to the same scaffold with at least 90% of their total length (Table S14). Three methods, including similarity-based, ab initio methods and genome-guided assembly of RNA-Seq, were applied to identify protein-coding genes. After manual verification, a total of 13,634 gene models were retained as the final gene set. Detailed analysis of gene length, exon number per gene and gene density in C. sinensis showed similar patterns to those seen in both S. japonicum and S. mansoni (Table 2). Approximately 76.6% of the genes have homologs in the NCBI non-redundant database, and 58.8% can be classified using Gene Ontology terms. Overall, 79.6% of the genes could be annotated (Table S2). In addition, non-coding RNAs were identified by the methods mentioned in our previous study [1]. Nine rRNA fragments, 276 tRNAs, 482 small nucleolar RNAs, 149 small nuclear RNAs and 165 miRNA precursor genes were identified in the C. sinensis genome (Table S14), and 107 miRNAs had been found expressed previously [3] (Table S15). Methods of assembly and annotation DNA library construction and sequence analysis Besides paired-end data previously published [1], we sequenced two paired-end and two mate-pair libraries again. The two short-insert (400and 550-bp) DNA libraries were constructed from the same fluke used in our previous study [1] as described in the Paired-End Sample Preparation Guide (Illumina, San Diego, CA, USA). In addition, the two long-insert (2000- and 5000-bp) DNA libraries were constructed from twenty adult worms that were pooled together using the SOLiD Mate-Paired Library Construction Kit. Cluster generation was performed on the cBot (Illumina), following the cBot User Guide. A paired-end sequencing run was then performed on the Genome Analyzer IIx (Illumina) as described in the Genome Analyzer IIx User Guide (Illumina). After masking adaptor sequences, removing contaminated reads and trimming low-quality reads, the cleaned data were processed for computational analysis. Genome assembly and repeat identification The whole genome shotgun sequencing raw data were filtered using Fastx-tools [4][13] using the following criteria: 1) reads containing sequencing adaptors were removed; 2) reads were trimmed to 103bp for Celera Assembler limitation; 3) reads with low-quality (the percentage of nucletides with Q20 score were lower than 50%) were removed; and 4) artificial reads were removed. The Celera Assembler v6.1 [5] was used to assemble contigs and construct scaffolds with the mate-pair data using SSPACE [6]. Finally, the gaps were filled with GapCloser [7][16]. Known repetitive elements were identified using RepeatMasker [8,9] with the Repbase database [10,11] (version: 2009-06). A de novo repeat library was constructed using RepeatModeler [12,13], and default parameters were then used to generate consensus sequences and classification information for each repeat family. RepeatMasker was again run on the genome using the repeat library built with RepeatModeler. Gene prediction Predicted proteins from S. japonicum and S. mansoni were aligned to the C. Sinensis transcriptome to identify conserved genes. Because the GeneWise [14] program is time consuming, proteins from schistosome were first aligned with the C. sinensis genome using genBlastA [15]. Subsequently, matched genomic regions were extracted and GeneWise was used to identify exon/intron boundaries. The Program to Assemble Spliced Alignments (PASA) [16] was used to generate spliced alignments of putative full-length cDNAs to the unmasked assembly, which was then used to train the ab initio gene prediction software, Augustus [17]. Genscan [18] was run using the model parameters for human. The RNA-Seq data from the four tissues were aligned to the genome using TopHat [19]. Cufflinks [20] was then used to assemble the transcripts with junction information. Gene predictions were generated using Augustus and Genscan, and spliced alignments of S. japonicum and S. mansoni proteins and transcripts produced by Cufflinks were integrated with EvidenceModeler [21]. Transcript isoforms were constructed by Cufflinks with -g and default parameters, followed by Cuffcompare. Alternative splicing events were predicted using altSpliceFinder program in Ensembl API tools. Protein domain analysis InterProScan [22] was run on all species (C. sinensis, S. japonicum, S. mansoni, C. elegans, D. melanogaster, D. rerio, G. gallus and H. sapiens) to predict protein sequences. Matched sequences tagged as ‘True Positive’ (status ‘T’) by InterProScan were retained. Functional annotation C. sinensis reference genes were mapped to KEGG pathways [23] by BLAST (e-value<1e-5). BLAST searches against the Swiss-Prot database and NCBI non-redundant database (e-value<1e-5) were conducted to provide comprehensive functional annotation. CEGMA validation Using default parameters, the CEGMA [2] set of 458 core eukaryotic genes was used to evaluate the completeness of the predicted gene models using the GenBlastA [15] program. References 1. Wang X, Chen W, Huang Y, Sun J, Men J, et al. (2011) The draft genome of the carcinogenic human liver fluke Clonorchis sinensis. Genome Biol 12: R107. 2. Parra G, Bradnam K, Korf I (2007) CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23: 1061-1067. 3. Xu MJ, Liu Q, Nisbet AJ, Cai XQ, Yan C, et al. (2010) Identification and characterization of microRNAs in Clonorchis sinensis of human health significance. BMC Genomics 11: 521. 4. Fastx-tools website. Available: http://hannonlab.cshl.edu/fastx_toolkit/.Accessed 2012 August 25. 5. Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, et al. (2000) A whole-genome assembly of Drosophila. Science 287: 2196-2204. 6. Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W (2011) Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27: 578-579. 7. GapCloser website. Available: http://soap.genomics.org.cn/about.html. Accessed 2012 August 25. 8. Tarailo-Graovac M, Chen N (2009) Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4: Unit 4 10. 9. Chen N (2004) Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4: Unit 4 10. 10. Kapitonov VV, Jurka J (2008) A universal classification of eukaryotic transposable elements implemented in Repbase. Nat Rev Genet 9: 411-412; author reply 414. 11. Kohany O, Gentles AJ, Hankus L, Jurka J (2006) Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics 7: 474. 12. Levitsky VG (2004) RECON: a program for prediction of nucleosome formation potential. Nucleic Acids Res 32: W346-349. 13. Price AL, Jones NC, Pevzner PA (2005) De novo identification of repeat families in large genomes. Bioinformatics 21 Suppl 1: i351-358. 14. Birney E, Clamp M, Durbin R (2004) GeneWise and Genomewise. Genome Res 14: 988-995. 15. She R, Chu JS, Wang K, Pei J, Chen N (2009) GenBlastA: enabling BLAST to identify homologous gene sequences. Genome Res 19: 143-149. 16. Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith Jr RK, et al. (2003) Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Research 31: 5654. 17. Stanke M, Waack S (2003) Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19 Suppl 2: ii215-225. 18. Burge C, Karlin S (1997) Prediction of complete gene structures in human genomic DNA. J Mol Biol 268: 78-94. 19. Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105-1111. 20. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, et al. (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511-515. 21. Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, et al. (2008) Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9: R7. 22. Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, et al. (2005) InterProScan: protein domains identifier. Nucleic Acids Res 33: W116-120. 23. Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28: 27-30.