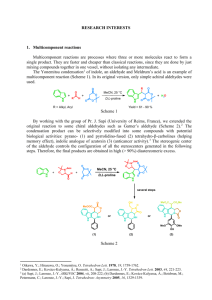
Microsoft Word
advertisement

Abstract The thesis entitled “Synthesis of C-Linked Carbo-3, 4-Amino Acids, Peptides and Studies Towards the Synthesis of Marinomycin A” is divided into three chapters. Chapter I is dealt with the synthesis of new mixed -peptides from the -Caa monomers having D-xylose, D-lyxose, D-ribose and D-arabinose side chains, to understand the impact of the side chains. Chapter II is divided into two sections, wherein Section A is dealt with the synthesis of new amino acid and its conversion into /-peptides to realize 12/10-helices, while, the Section B is dealt with the synthesis of new bifunctional -amino acid and its conversion into mixed peptides to realize 9-helix. Chapter III is dealt with the attempted synthesis of marinomycin A. Chapter I: Synthesis of Mixed -Peptides from C-Linked Carbo--Amino Acids (-Caas) and Study on the Impact of Carbohydrate Side chain. Carbohydrates as well as peptides and proteins are essential biopolymers of life. To perform biological functions, correct folding of biopolymers which creates active site is crucial1. β-Amino acids are much less frequent in nature than α-amino acids and have also been found incorporated into naturally occurring peptides with important pharmacological properties2. Seebach et al.3 and Gellman et al.4 were the first to report β-peptides with novel helical structures.5 Based on the structures of nikkomycins6, our group reported the synthesis of Clinked carbo-β-amino acids7 (β-Caa), with carbohydrate moieties as side chains and converted them into β-peptides8 with 12/10-helical patterns. This chapter is dealt with the synthesis of new mixed carbo--peptides from -Caas having D-xylose, D-mannose, D-ribose and D-arabinose side chains. The main idea behind the study is to understand the sterochemical as well as other structural features of the carbohydrate side chains that are essential for minimum compatibility to adopt well-defined helices in the mixed peptides. Thus, the requisite -Caa monomers 1-6 were prepared by aza-Michael addition with benzyl amine on the esters derived from the corresponding aldehdyes obtained from D-glucose, Dmannose, D-ribose and D-arabinose (Scheme 1). Thus, reaction of the esters 7 with benzyl amine gave the esters 8 and 9 as separable mixtures of diastereoisomers. Hydrogenolysis of 8 and 9 followed by reaction of the respective amines with Boc2O and Et3N in THF afforded the -Caa monomers 1-6. O O NHBoc O H3CO O O O H3CO O 1 [Boc-(S)--Caa(x)-OMe] H3CO O 2 [Boc-(R)--Caa(x)-OMe] O NHBoc H3CO OMe O 4 [Boc-(R)--Caa(l)-OMe] O O MeO 5 [Boc-(S)--Caa(r)-OMe] O O O H3CO O NHBoc O NHBoc OMe O OMe H3CO 3 [Boc-(S)--Caa(l)-OMe] O O O NHBoc O O MeO O MeO NHBoc 6 [Boc-(S)--Caa(Da)-OMe] Scheme 1 NHBn O O MeO BnNH 2 R 7 MeO R O + R= MeO O O R Boc2O, Et3N, THF 9a-c O O Pd-C, MeOH 1-6 MeO 8a-d O NHBn OMe O O MeO O O O O OMe O Mixed -peptides from (R)-β-Caa(x) and (S)-β-Caa(l) with D-xylose and D-lyxose side chains Based on the molecular mechanics calculations of Wu and Wang,9 and from the earlier studies of our group, in the present study on the synthesis of mixed-β-peptides from β-Caa(x) 2 and β-Caa(l) 3, it was planned to adopt the same stratergy of alternating chirality. Both the side chains are furanoside in nature and in D-form, while, the acetonide groups are in 1,2- and 2,3postions and are in α-and β-face in 2 and 3 respectively. Synthesis of mixed-β-peptides is based on the 1:1 alternating use of (R)-β-Caa(x) 2 and (S)-β-Caa(l) 3 (Scheme 2). The peptides were prepared using standard peptide coupling conditions in the presence of EDCI and HOBt, with DIPEA as a base in dry CH2Cl2. Accordingly, the dipeptide 10, tetrapeptide 11 and hexapeptide 12 were prepared by adopting segment condensation method. NMR, CD and MD studies clearly indicated the presence of novel right-handed 10/12-mixed helical pattern in the mixed--peptides 11 and 12 (Scheme 2). Mixed-β-peptides from (R)-β-Caa(x) and (S)-β-Caa(r) with D-xylose and D-ribose side chains In a further study to understand the impact of the carbohydrate side chains, a new design of a mixed peptide was proposed wherin the β-Caa(x) with D- xylose as side chain will remain the same, while, -Caa(l) with D-lyxose side chain is replaced with -Caa(r) with D-ribose as side Scheme 2 H N O H N O O O H3CO O 12 H N O O O O O O O O O OCH3 12 H N O O 10: n = 1 dipeptide 11 n = 2 tetrapeptide 12: n = 3 hexapeptide O O 10 H N H3CO OCH3 n 10 H N H N O O O O O O H3CO O OCH3 O H N O O H3CO O OCH 3 O OCH3 O O O O O OCH 3 O 12 chain, wherein, the sterocentres at C-1, C-2 and C-3 are enantiomeric to -Caa(l). Accordingly, the peptides 13-15 were prepared from (R)-β-Caa(x) 2 and (S)-β-Caa(r) 5 in the presence of EDCI and HOBt, with DIPEA as a base in dry CH2Cl2 (Scheme 3). The NMR, CD and MD studies of peptides 14 and 15 clearly indicated the presence of robust 12/10-mixed helix (Scheme 3). Scheme 3 H N O H N O O O H3CO O 12 H N O H N O 10 O O O O OCH3 12 H N O O O H3CO O OCH O 3 13: n = 1 dipeptide 14: n = 2 tetrapeptide 15: n = 3 hexapeptide O O O O O H3CO OCH3 n 10 H N H N O O O O O O O H N O H3CO OCH3 OCH3 O O O O O O O OCH3 15 Mixed-β-peptides from (R)-β-Caa(l) and (S)-β-Caa(Da) with D-lyxose and D-arabinose side chains From the above two designs using β-Caa(x) having a D-xylose side chain with two different β-Caas having D-lyxose and D-ribose side chains, which are enantiomeric at the C-1, C-2 and C-3 positions, the mixed peptides have shown the presence of 12/10-mixed helices. Thus, it indicated that in these systems the sugar side chain has no impact on helix formation. Hence, in a new design, it was proposed to use a different β-Caa side chain in place of D-xylose moiety to understand more about the side chain impact. In the new design, β-Caa(Da), having a Darabinose side chain, was used in place of β-Caa(x) monomer. The stereochemistry of the 1,2-Oisopropylidene group in β-Caa(Da) 6 is now inverted (in β-face) unlike in β-Caa(x) 1. Accordingly, the peptides 16-18 were prepared from (R)-β-Caa(l) 4 and (S)-β-Caa(Da) 6 in the presence of EDCI, HOBt and DIPEA in CH2Cl2 (Scheme 4). Scheme 4 H N O H N O OCH3 O O O O 16: n = 1 dipeptide 17: n = 2 tetrapeptide 18: n = 3 hexapeptide O H3CO O OCH3 O O n H N O H N O H N O O O O O H3CO OCH3 H N O O O H N O O O O H3CO OCH3 O O O H N O OCH3 O O O O H3CO OCH3 O O O O 18 Thus, even though we were successful in the synthesis of peptides having alternating βCaa(l) 4 and β-Caa(Da) 6, the spectral analysis revealed that they exist as rotamers in CDCl3 solutions. Due to the presence of rotamers always, the secondary structures could not be understood well in the NMR studies on these peptides. Thus, for the first time it has become evident that the compatibility of the side chain with appropriate sterochemistries is equally important for a robust helix formation in this class of β-peptides. Mixed-β-peptides from (R)-β-Caa(l) and (S)-β-Caa(r) with D-lyxose and D-ribose side chains From the above study it was well understood that the compatibility of the side chain is also an essential and important aspect in defining a proper secondary helical structure. To further understand the side chain impact, mixed β-peptides were proposed from alternating use of (R)-β- Caa(l) 4 and (S)-β-Caa(r) 5 with D-lyxose and D-ribose side chains, wherein, though both the side chains have 2,3-O-isopropylidene groups, the stereocentres at the C-1, C-2 and C-3 in both these monomers are enantiomeric to each other. Accordingly, the peptides 19-21 were prepared from (R)-β-Caa(l) 4 and (S)-β-Caa(r) 5 in the presence of EDCI, HOBt and DIPEA in CH2Cl2 (Scheme 5). Scheme 5 O H N H N O O O O H N O O H N O O O O OCH3 O OCH3 H N O O O OCH3 n H N O O 19: n = 1 dipeptide 20 n = 2 tetrapeptide 21: n = 3 hexapeptide O O O O OCH3 O OCH3 H N O O O OCH3 O H N O O OCH3 OCH3 O O O O OCH3 O O O O OCH3 21 In 1H NMR of 20 and 21, characteristic information on a well definded secondary structure was not observed. Further, solvent titration studies did not show their involvement in H-bonding. Thus, mixed -peptides were prepared utilizing the concept of ‘alternating chirality’ from -Caas 1-6 having D-xylose, D-lyxose, D-ribose and D-arabinose side chains. The spectral and analytical studies amply indicated that, all the side chains are not compatible in giving novel secondary structures. The structural features and enantiomeric nature of the carbohydrate side chain plays a role in defining the conformations in such peptides. Chapter II: Synthesis of New C-Linked Carbo-γ4-Amino Acids and Their use in the Synthesis of Peptides with Novel Helical Structures Section A: Synthesis of New C-Linked Carbo-γ4-Amino Acids (γ4-Caas) and their Conversion into /-Hybrid Peptides Most of the unnatural foldamers reported so far have homogeneous backbones i.e., they are constructed from single class of amino acids. Heterogeneous backbones composed of two or more residue types also display well-defined folding behavior, and such an exploration is important because mixing monomer classes leads to an exponential increase in the range of potential foldamers. Based on our earlier findings on /-hybrid peptides10 derived from (S)--Caa and L-Ala repeats with unprecedented 11/9-mixed helices and recent theoretical studies by Hofmann et al.11, we envisaged that /- hybrid peptides from (S)--Caa and L-Ala would adopt novel 10/12or 12/10-mixed helical structures in such peptides. Inspired by these findings, (S)--Caa(l), a Clinked carbo--amino acid 22 from D-mannose, was prepared by homologation method from the known -amino acid (Scheme 6). The new design on /-hybrid peptides from alternating L-Ala and (S)--Caa, would thus be the first motif devoid of β-amino acid for a 12/10-mixed helix. Scheme 6 BocHN BocHN OCH3 O O O BocHN OCH3 O O O O BocHN O OCH3 25 O + d OCH3 (After separation of -monomer) O O BocHN O O O 24 OCH3 O O 3 OCH3 O 23 N2 O OEt O OCH3 O 3 BocHN O b O O c OH a OCH3 O O O OCH3 22 Reactions and reagents: a) aq. 4N NaOH; MeOH, 0 oC-RT; (b) ClO2CEt, Et3N, -25oC-RT; (c) CH2N2 , Ether, KOH, -25 oC-RT ; (d) Et3N, PhCO2Ag,.-25 oC, 8 h. Accordingly, the di-, tetra- and hexapeptides 26-28 were prepared from alternating (S)-γCaa 22 and L-Ala in the presence of EDCI, HOBt and DIPEA in CH2Cl2 (Scheme 7). NMR, CD and MD studies clearly indicated the presence of novel right-handed 12/10-mixed helical pattern in the γ/-hybrid peptides 27 and 28 (Scheme 7). Scheme 7 H N O O OCH3 N H O O 26: n = 1 dipeptide 27: n = 2 tetrapeptide 28: n = 3 hexapeptide O O O OCH3 n 12 12 10 O H N O N H O OCH 3 N H O O O O O O OCH 3 O H N N H O O O O O O H N OCH 3 O OCH 3 28 Similarly, new /γ-peptides 29-31 (Scheme 8) with L-Ala at the N-terminus were also prepared from L-Ala and (S)-γ-Caa 22 under standard peptide coupling conditions (EDCI, HOBt and DIPEA) in CH2Cl2. NMR, CD and MD studies evidently proved the presence of a righthanded 12/10-mixed helical pattern in /γ-hybrid peptides 29-31. Scheme 8 O O N H O H N OCH3 N H O H3CO O 29: n = 1 tripeptide 30: n = 2 pentapeptide 31: n = 3 heptapeptide O O O n 12 12 10 O O N H 10 O H N O N H O O O 12 OCH 3 O H N O O N H O O H N O N H O OCH 3 O O O OCH 3 O OCH3 Section B: Synthesis of New Bifunctional C-Linked Carbo-γ4-Amino Acid and Conversion into Mixed -Peptides Using Alternating -Caa and GABA Based on our earlier findings8b on mixed β-peptides and -peptides derived from alternating (S)-β-Caa/β-hGly and -Caa(x)/GABA, resulting in left-handed 12/10-mixed helices 9-helix,12 respectively, a new bifunctional (S)--Caa(l) 32 was proposed. The new (S)--Caa(l) 32 was prepared by homologation of the bifunctional -amino acid prepared from D-mannose (Scheme 9). The new (S)--Caa(l) 32 would not only result in the 9-helix in the mixed peptides with GABA, but also would help in the solubility in water, that would enhance the possibility for biological evaluation. Using -(S)-Caa(l) 32, previously reported 4-(S)–Caa and GABA, di-, tetra- and hexapeptides 36-38 under standard peptide coupling conditions (EDCI, HOBt and DIPEA) in CH2Cl2 were prepared. NMR and CD studies confirmed the presence a left-handed 9-helix in tetrapeptide 37. Though the presence of some conformation is indicated in CD of 38, it could not be deducted from NMR due to severe overlapping of the signals (Scheme 10). Scheme 9 O NHCbz O HO O NHBoc O EtO O O O O NHCbz O 35 O NHBoc b O NHCbz O H3CO c + 33 NHBoc O 34 33 N2 NHCbz O a NHBoc O O O O 32 o Reactions and reagents: (a) ClO2CEt, Et3N,-25 C-RT, (b) CH2N2, Ether, KOH; (c) Et3N, PhCO2Ag,.-25 oC-RT, 8 h Scheme 10 9 O O H N O O N H O O NHBoc O 36 O O O N H O O O H N O O O O HN OCH3 O O O OCH3 9 N H O O O OCH3 O 9 37 O H N O H N N H O O O O H N O OCH3 38 N H O H N O O O O OCH3 N H OCH3 O OCH3 O Chapter III. Attempts Directed Towards the Synthesis of Marinomycin A Marinomycin A 39, a macrodiolide, isolated from a novel marine Actinomycete13, (Marinispora strain CNQ-140), exhibited potent antibiotic activity against methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus faecium (VREF), with MIC90 values ranging from 0.1 to 0.6 µM. Marinomycin A 39 is a C2 symmetric14 system and arose great interest to the organic chemists.15a,b The retro analysis of 39 indicates that it can be prepared from the dimerisation of 40, which in turn could be envisaged from the fragments 41-43 (Scheme 11). Scheme 11 OH HO O O R S S S OH OH OH OH OH OH S S S S R O O OH 39 S OH HO O HO OH OH OH OH 40 OH O OR1 OR2 OR1 OR1 O + 41 O H 42 + O Ph S O O O 43 Synthesis of fragment 41 is achieved as shown in Scheme 12. The chiral epoxide 44,on reaction with 45, using n-BuLi, at -78 oC gave the alcohol 46 in 64% yield, which was protected as MOM ether using MOMCl and DIPEA in CH2Cl2 at 0 oC to offord 47 in 93% yield. Compound 47 was selectively deproteced using cat. PTSA in MeOH to give primary alcohol 48 in 87% yield, which on readuction with Red-Al in THF at 0 oC afforded trans allylic alcohol 49 in 91% yield. To generate the next chiral centre, this allylic alcohol was subjected to epoxidation under Sharpless epoxidation conditions using (+)-DIPT, Ti(i-OPr)4 and cumene hydroperoxide at -20 oC to obtain the chiral epoxide 50 in 77% yield. Epoxide 50 was then selectively opened as 1,3-diol using Red-Al in THF in 85% yield, which was subsequently protected using PMB acetal and cat. PTSA as the benzylidene derivative 52 in 67% yield. The acetal 52 was regioselectively opened into primary alcohol 53 with DIBAL-H in dry CH2Cl2, which on oxidation with DessMartin periodinane at 0 oC gave aldehyde 54 in 96% yield. Witting olefination of 54 gave the Scheme12 O OH OTHP a + 44 OTHP 45 OMOM b OTHP 46 OMOM 47 OR 1 OR 1 d OH O e OH f OH R1 = MOM 49 48 c 50 R1= MOM terminal olefin 55. in 72% yield. The olefin on epoxidation with m-CPBA in dry CH2Cl2 furnished the racemic epoxide 56 in 85% yield, which on Jacobsen Kinetic resolution using (R, R)-Jacobsen catalyst afforded the chiral epoxide 41 (C1-C7 fragment with three out of five stereocentres) in 43% yield. O OR1 OH OH g 51 R1= MOM OR1 O 52 k 55 R1= MOM h OR1 OPMB OR1 OPMB i OH OR1 OPMB j O R1= MOM OR1 OPMB O 56 R1= MOM O 53 R1= MOM l 54 R1= MOM OR1 OPMB O 41 Reagents and conditions: (a) n-BuLi, BF3.Et2O ,THF, -78 oC, 3 h; (b) MOMCl, DIPEA, CH2Cl2, 6 h; (c) PTSA, MeOH, 6 h; (d) Red-Al, THF, 3 h, 0 oC-RT; (e) (+)-DIPT, Ti(i-Opr)4, Cumene hydroperoxide,, CH2Cl2, 5 h, -20 oC; (f) Red-Al, THF, 4 h, 0 oC-RT; (g) PMB-acetal, CH2Cl2, cat. PPTS, 4 h, 0 oC-RT; (h) DIBAL-H, CH2Cl2, 6 h, 0 oC-RT; (i) DMP, CH2Cl2, 3 h, 0 oC-RT; (j) PPh3=CH2,K+ t--OBu, THF, 0 oC, 6 h; (k) m-CPBA, CH2Cl2, 8 h; (l) (R,R)-Jacobsen's Catalyst, AcOH, H2O, Tolune, 16 h. Synthesis of 42 was initiated from propargyl alcohol (Scheme 13). The allylated product 58 was subjected to epoxidation using m-CPBA in CH2Cl2 to afford the racemic epoxide 59 in 92% yield, which was resolved by Jacobsen Kinetic resolution using (S,S) Jacobsen catalyst to Scheme13 b a HO SPTO 57 d SPTO c SPTO OH e O SPTO O 60 O 61 SPTO O 59 58 OR2 62 O X O f SPTO R2 = MOM OR2 O OEt 63 R2= MOM Reagents and conditions: (a) TPSCl, Imidazole, Dry CH2Cl2, 6 h, 0 oC-RT; (b) m-CPBA, CH2Cl2, 8 h; (c) (S,S) Jacobsen catalyst; (S,S)-Jacobsen's Catalyst, AcOH, H2O, Tolune, 16 h; (d) Ethyl propiolate, n-BuLi, -78 oC, 3 h; (e) MOMCl, DIPEA, CH2Cl2, 6 h; (f) Me2CuLi, dry ether, -78 oC, 8 h. afford the chiral epoxide 60 in 44% yield. The epoxide 60 was treated with ethyl propiolate in presence of n-BuLi at -78 oC to give the ester 61 in 84% yield, which on reaction with MOMCl and DIPEA in CH2Cl2 at 0 oC furnished 62 (C8-C16 framework with one stereocentre) in 92% yield. Several attempts to functionalize 62 met with failure.