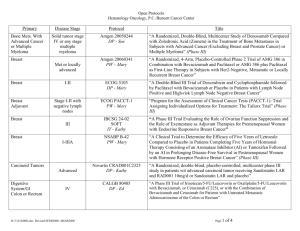
GOG-0241 - the Gynecologic Cancer InterGroup
advertisement

PROTOCOL GOG-0241 A GCIG INTERGROUP MULTICENTRE PHASE III TRIAL OF OPEN LABEL CARBOPLATIN AND PACLITAXEL +/- BEVACIZUMAB COMPARED WITH OXALIPLATIN AND CAPECITABINE +/- BEVACIZUMAB AS FIRST LINE CHEMOTHERAPY IN PATIENTS WITH MUCINOUS EPITHELIAL OVARIAN CANCER (MEOC) POINTS: PER CAPITA MEMBERSHIP - STUDY CHAIR DAVID M. GERSHENSON, M.D. M.D. ANDERSON CANCER CTR DEPARTMENT OF GYN/ONC 1515 HOLCOMBE BLVD., BOX 440 HOUSTON, TX 77030-4009 (713) 792-2455 FAX: (713) 792-7586 E-MAIL: dgershen@mdanderson.org STUDY CO-CHAIR RICHARD T. PENSON, M.D. MASSACHUSETTS GENERAL HOSPITAL 55 FRUIT STREET, YAWKEY 9066 BOSTON, MA 02114-2617 (617) 726-5867 FAX: (617) 724-6898 EMAIL: rpenson@partners.org NURSE CONTACT JACALYN GANO M.D. ANDERSON CANCER CTR DEPARTMENT OF GYN/ONC P.O. BOX 301439 HOUSTON, TX 77230-1439 (713) 794-1422 FAX: (713) 792-7586 E-MAIL: jgano@mdanderson.org PATHOLOGIST RICHARD ZAINO, M.D. (SEE GOG WEBSITE DIRECTORY) TRANSLATIONAL CO-CHAIR JEFF BOYD, Ph.D. (SEE GOG WEBSITE DIRECTORY) TRANSLATIONAL CO-CHAIR ANIL SOOD, M.D. (SEE GOG WEBSITE DIRECTORY) PATHOLOGIST KAREN E. STREHLOW, M.D. (SEE GOG WEBSITE DIRECTORY) QUALITY OF LIFE CO-CHAIR MICHAEL FRUMOVITZ, M.D. (SEE GOG WEBSITE DIRECTORY) STATISTICIAN WILLIAM E. BRADY, M.S. (SEE GOG WEB SITE DIRECTORY) TRANSLATIONAL RES. SCIENTIST ZOE MINER, PH.D. (SEE GOG WEB SITE DIRECTORY) Draft: July 9, 2009 This protocol was designed and developed by the Gynecologic Oncology Group (GOG). It is intended to be used only in conjunction with institution-specific IRB approval for study entry. No other use or reproduction is authorized by GOG nor does GOG assume any responsibility for unauthorized use of this protocol. PROTOCOL GOG-0241 A GCIG INTERGROUP MULTICENTRE TRIAL OF OPEN LABEL CARBOPLATIN AND PACLITAXEL +/- BEVACIZUMAB COMPARED WITH OXALIPLATIN AND CAPECITABINE +/- BEVACIZUMAB AS FIRST LINE CHEMOTHERAPY IN PATIENTS WITH MUCINOUS EPITHELIAL OVARIAN CANCER (MEOC) Schema mEOC Eligible patients aged 18 or over with newly diagnosed advanced mucinous ovarian carcinoma FIGO stage II – IV OR recurrent stage I Randomize N=322¶ Carboplatin AUC 6 Paclitaxel 175mg/m2 6 cycles† Each cycle =21 days N=83 Oxaliplatin 130 mg/m2 Capecitabine 850mg/m2 bd 6 cycles† Each cycle =21 days† N=83 Clinical assessments every 3 months for 36 weeks Telephone assessments every 3 weeks for 36 weeks Treatment to start within 14 days of randomization for all arms Carboplatin AUC 6 Paclitaxel 175mg/m2 Bevacizumab 15 mg/kg 6 cycles†‡ Each cycle =21 days† N=83 Oxaliplatin 130 mg/m2 Capecitabine 850mg/m2 bd Bevacizumab 15 mg/kg 6 cycles†‡ Each cycle =21 days N=83 Bevacizumab 15 mg/kg 12 cycles† Each cycle= 21 days Clinical assessments every 6 weeks for 36 weeks (i.e., the twelve 21-day cycles) Follow-up: Every 3 months for 2 years Every 6 months for years 3-5 ¶ Approximately 60 (or about 20%) of the patients are expected to be enrolled by GOG sites. † Or until progression of disease or adverse effects prohibit further therapy. ‡ Bevacizumab can be omitted from the first cycle if chemotherapy must be started within 4 weeks of surgery. Please refer to Section 5 for further details. TABLE OF CONTENTS PAGE 1.0 OBJECTIVES 1 2.0 BACKGROUND AND RATIONALE 2 3.0 PATIENT ELIGIBILITY AND EXCLUSIONS 4 4.0 STUDY MODALITIES 7 5.0 TREATMENT PLAN AND ENTRY/RANDOMIZATION PROCEDURE 21 6.0 TREATMENT MODIFICATIONS 30 7.0 STUDY PARAMETERS 39 8.0 EVALUATION CRITERIA 44 9.0 DURATION OF STUDY 52 10.0 STUDY MONITORING AND REPORTING PROCEDURES 53 11.0 STATISTICAL CONSIDERATIONS 58 12.0 BIBLIOGRAPHY 64 SUGGESTED PATIENT INFORMATION/INFORMED CONSENT APPENDIX I - Clinical Staging (FIGO) APPENDIX II - New York Heart Association Criteria APPENDIX III- Peripheral Vascular Disease APPENDIX IV- Anaphylaxis Precautions APPENDIX V - Patient Pill Calendar APPENDIX VI- Blood Pressure Check Calendar -11.0 GOG-0241 OBJECTIVES 1.1 1.2 1.3 Primary Objective 1.11 To determine if capecitabine and oxaliplatin reduces the death rate compared to carboplatin and paclitaxel in women with mucinous adenocarcinoma of the ovary or fallopian tube. 1.12 To determine if bevacizumab reduces the death rate compared to no bevacizumab in women with mucinous adenocarcinoma of the ovary or fallopian tube. Secondary Objectives 1.21 To determine if capecitabine and oxaliplatin increases the duration of progression-free survival (PFS) compared to carboplatin and paclitaxel in women with mucinous adenocarcinoma of the ovary or fallopian tube. 1.22 To determine if bevacizumab increases the duration of PFS compared to no bevacizumab in women with mucinous adenocarcinoma of the ovary or fallopian tube. 1.23 To compare the response rates for capecitabine and oxaliplatin versus carboplatin and paclitaxel in patients with mucinous adenocarcinoma of the ovary or fallopian tube with measurable disease after initial tumor reductive surgery. 1.24 To compare the response rates for bevacizumab versus no bevacizumab in patients with mucinous adenocarcinoma of the ovary or fallopian tube with measurable disease after initial tumor reductive surgery. 1.25 To determine the nature and degree of toxicity of capecitabine and oxaliplatin compared with that of carboplatin and paclitaxel in this cohort of patients. 1.26 To determine the nature and degree of toxicity of bevacizumab in this cohort of patients. 1.27 To compare capecitabine and oxaliplatin versus carboplatin and paclitaxel with respect to changes in patient reported neurotoxicity. 1.28 To determine the impact on Quality of Life (QOL, as measured by the FACT-O TOI) following treatment with the above regimens Exploratory Objectives 1.31 To identify expression of VEGF ligand, receptor, and activated receptor via immunohistochemistry in mucinous adenocarcinoma of the ovary. 1.32 To identify expression of EGF ligand, receptor, and activated receptor via immunohistochemistry in mucinous adenocarcinoma of the ovary. -2- 1.33 2.0 GOG-0241 To assess the mutational status of the KRAS oncogene in primary mucinous ovarian carcinomas obtained prior to treatment with oxaliplatin/capecitabine and then to determine whether there exists evidence for a correlation between the presence of KRAS mutation and response to therapy in this group of ovarian cancer patients. BACKGROUND AND RATIONALE There will be an estimated 20,180 new cases of ovarian cancer in the United States in 2006, but only 9% of these will have mucinous histology.1,2 Overall, survival of women with mucinous adenocarcinoma of the ovary is better than its serous counterpart, but this is likely owing to the fact that at diagnosis, mucinous carcinomas are often confined to the ovary, while serous carcinomas are usually metastatic. In fact, over half of mucinous adenocarcinomas are localized to the ovary at surgery, whereas over 80% of serous adenocarcinomas have distant spread.3 However, women with advanced-stage mucinous carcinomas of the ovary have a much shorter progression-free and overall survival than those with advanced-stage serous carcinomas and are more likely to fail adjuvant platinum-based chemotherapy regimens.4 In addition to differences in clinical behavior, laboratory data support molecular differences between the different epithelial subtypes of ovarian cancer. When gene expression arrays were performed on the subtypes of epithelial ovarian cancers, mucinous cancers grouped separately from serous, endometrioid, and clear-cell tumors on dendrograms.5 In addition, KRAS more often overexpressed in mucinous tumors when compared to serous, endometrioid, or clear-cell tumors.6 The Ras family of proteins consists of three isoforms (H-, K-, and N-Ras) that play a critical role in controlling normal and malignant cell growth7. KRAS mutation is one of the most common abnormal genetic events in human cancer, with the highest incidence in pancreatic carcinomas (90%) and colorectal tumors (50%).8 Mitogen-activated protein kinases (MAPKs) are the best-characterized signal pathways in transduction and regulation of Ras activity and cellular proliferation. Phosphorylation of its membrane-anchoring domain by protein kinase-C (PKC) drives KRAS off the plasma membrane and onto the outer mitochondrial membrane where it induces cell death. Traditionally, women with all ovarian epithelial carcinomas have been treated similarly, with upfront tumor debulking surgery followed by platinum-based adjuvant chemotherapy. Support for this approach comes from Gynecologic Oncology Group (GOG) collaborative trials,9,10 as well as studies in Europe and Canada.11 These studies showed platinum/taxane-based therapies to have an advantage in women with advanced-stage epithelial ovarian cancers. However, only 3 to 4% of patients in each of those studies had mucinous histologies and, owing to small numbers, subset analysis of these women has not been possible. Unlike the 26% response rate reported for platinum-based chemotherapy regimens in patients with mucinous ovarian carcinomas,4 combination therapy with a fluorouracil and either CPT-11 (irinotecan) or oxaliplatin have response rates of 50% in patients with advanced-stage colorectal cancers.12,13 In one study of patients with advanced mucinous colorectal cancer, subjects treated with a combination of 5-FU and oxaliplatin (FOLFOX) had a better response rate, longer overall survival, and fewer chemotherapy-related side effects than those treated either with CPT-11/5-FU or CPT-11/oxaliplatin combination therapies.14 More recently, Phase I/II studies of the combination of capecitabine (Xeloda) and oxaliplatin (XELOX) as first-line therapy have shown this combination to be as effective as the FOLFOX regimen.15,16 The GOG -3- GOG-0241 has evaluated oxaliplatin alone in platinum-resistant epithelial tumors of the ovary, with minimal activity shown.17 However, of the 23 evaluable patients, none had mucinous histology. Mucinous ovarian carcinomas seem to have a clinical course that is more similar to mucinous gastrointestinal carcinomas than to serous ovarian carcinomas. We hypothesize that multimodality therapy with capecitabine and oxaliplatin (XELOX) will offer a better response, longer progression-free survival, and longer overall survival than current platinum/taxane strategies in patients with mucinous ovarian cancers. This trial will help determine if oxaliplatin and capecitabine +/- bevacizumab improves OS and PFS when compared to carboplatin and paclitaxel +/- bevacizumab in women with advanced stage mucinous ovarian cancer. Acute and chronic toxicities from carboplatin and taxol and their effect on QOL in women with gynecologic cancers is well-documented. 18-20 The effects on QOL by the addition of bevacizumab to this regimen for women with gynecologic malignancies is largely unknown. The effects on QOL with the oxaliplatin and capecitabine +/bevacizumab regimen in women with ovarian cancer has never been explored. Although data on QOL with these regimens exist for patients with colorectal cancer, 21-22 a direct comparison of QOL in patients receiving oxaliplatin and capecitabine vs oxaliplatin, capecitabine and bevacizumab, however, does not exist. Although high-quality, comparative QOL data for these regimens do not exist, we do have a number of studies from which we might examine regimen toxicities. GOG 182-ICON 5 enrolled over 4300 women into a 5-arm, randomized study with carboplatin (AUC 6) and paclitaxel (175 mg/m2) given every three weeks as one of the regimens. In this study, 864 evaluable patients received this combination. In this cohort, 24% had grade 2-4 peripheral neuropathy. 23 Peripheral neuropathy, whose symptoms include parasthesias of the hands and feet, can be severe with the carboplatin and paclitaxel combination and often is the dose limiting toxicity of the regimen. Paclitaxel is the main source of neuropathy in women receiving this regimen and its effects are cumulative. Most women have some resolution of symptoms after discontinuation of therapy but significant peripheral neuropathy, often described as “stocking/glove distribution” may be permanent in a small percentage of patients. Myelosuppression was a more significant toxicity as 59% had grade 4 neutropenia and 22% had a grade 3 or 4 thrombocytopenia.23 In this study, however, subjects received 8 cycles of the carboplatin/paclitaxel regimen. A phase II study of capectibine and oxaliplatin in advanced or metastatic colorectal cancer revealed significant (grade 3 or 4) diarrhea in 35% of the patients who had not been pretreated. 24 In this study, however, diarrhea was greatly reduced to < 15% when the capecitabine was reduced by 25% after the first episode of grade 3 or 4 diarrhea. In addition, 16% had “sensitive” neuropathy, 5% had nausea and vomiting, and 12% had thrombocytopenia.24 In a recent study of oxaliplatin, capecitabine and bevacizumab in patients with metastatic colorectal cancer, 73% of patients experienced grade 3 or 4 toxicity22. This included hand-foot reaction (19%), diarrhea (19%), hypertension (14%) fatigue (13%), sensory neuropathy (10%), nausea/vomiting (8%), and venous thromboembolic disease (7%). In this study, however, the capecitabine was administered at 100 mg/m2 BID which is higher than in the current protocol. The neurotoxicity most often reported from this combination is secondary to administration of the oxaliplatin. Oxaliplatin neuropathies may be either acutely with drug administration or chronic from cumulative effects of multiple doses. The acute syndrome typically consists of burning, numbness and/or tingling in the oropharyngeal cavity. This acute phase is almost always transient with resolution within hours or days after drug administration. The chronic neuropathy is similar to that of paclitaxel presenting as parasthesia of the hands and feet.25 Similar to the peripheral neuropathy experienced after paclitaxel -4- GOG-0241 administration, most women with parasthesia from oxaliplatin will have resolution of symptoms when the chemotherapy is discontinued although some might have chronic, debilitating neuropathy. In the current trial, QOL will be assessed using the Trial Outcome Index of the Functional Assessment of Cancer Therapy-Ovary (FACT-O TOI).26,27 This 26-item summary score captures the FACT-G QOL dimensions of Physical Well-Being (7 items), Functional WellBeing (7 items), and the Ovarian Cancer Subscale (12 item). By combining these three subscales, one is assured of capturing the full range of physical aspects of QOL in advanced ovarian cancer, including pain, fatigue, abdominal symptoms and functional status. By combining questions GP4, O1, and O3, which assess abdominal pain, swelling and cramps respectively, a comprehensive patient reported assessment of disease related abdominal symptoms including ascites can be evaluated. The neuropathic side effects of therapy will be measured by patient reported neurotoxicity, using: (1) FACT-GOG/NTX4, which is a 4-item scale that reliably and validly assesses platinum/paclitaxel-induced peripheral neuropathy, with Cronbach’s alpha ranging from 0.80 to 0.8528 and includes questions regarding numbness and tingling in hands and feet and discomfort in hands and feet; and (2) a question regarding cold-induced pain in the extremities, which can result from treatment with oxaliplatin. The neurotoxicity assessment will be administered prior to receiving the first cycle of therapy, just before starting the fourth cycle of chemotherapy, after the final cycle of chemotherapy, and one year after completing the final cycle of chemotherapy regardless if the ovarian cancer has remained in remission or has recurred. Although we may know many of the toxicities associated with these chemotherapy regimens, we do not know their effects on QOL. We hypothesize that although oxaliplatin and capecitabine +/- bevacizumab will improve OS and PFS when compared to carboplatin and paclitaxel +/- bevacizumab, the acute and chronic toxicities of the oxaliplatin/capecitabine regimen may affect QOL more adversely than carboplatinum/paclitaxel regimen. These data may prove invaluable both for patient education prior to initiating therapy as well as for helping physicians and patients choose the best treatment regimen for them. In addition to traditional chemotherapeutics, bevacizumab (Avastin), a monoclonal antibody to vascular endothelial growth factor (VEGF), has improved outcomes in patients with colorectal cancer. For untreated colorectal cancers, the combination of 5-FU with bevacizumab significantly improved response rate, progression-free interval, and overall survival when compared to 5-FU alone.29 Furthermore, in a randomized phase III trial in which patients were randomized in a 2 x 2 factorial design to receive either XELOX or FOLFOX-4, and then to bevacizumab or placebo, the addition of bevacizumab significantly improved PFS, but there was no difference in OS.30 This agent should have potent activity in mucinous carcinomas of the ovary, as studies have shown high levels of VEGF production by these tumors.31,32 Multiple phase II studies of bevacizumab in recurrent epithelial ovarian cancer have revealed response rates in the range of 16% to 24%.33-35 Recent findings suggest that the VEGF receptors are present on the ovarian cancer cells as well as being present on the tumor-associated endothelial cells.36 Specifically, VEGFR-2 was the predominant receptor noted on ovarian cancer cells. Therefore, it is possible that VEGFtargeted therapy may affect both the tumor vasculature as well as the tumor cells. While -5- GOG-0241 previous studies have attempted to relate the presence of ligand with response to therapy, data regarding the presence of total and activated VEGF-receptors on the tumors cells are largely lacking. Thus, the studies proposed here will provide new information regarding VEGF and EGF receptors on mucinous ovarian cancers. Other monoclonal antibodies such as cetuximab (Erbitux) target the epidermal growth factor receptor (EGFR). Although there has been mixed reporting of EGF ligand and receptor expression in epithelial ovarian cancers, there is support in the literature for the idea that mucinous tumors express EGF and EGFR37,38 which could be potential targets for biologic agents in the treatment of advanced-stage mucinous tumors. Also, activating mutations of the KRAS oncogene, specifically affecting codon 12, are observed in approximately 50% of invasive epithelial ovarian carcinomas of mucinous histology, but rarely in ovarian cancers of other histologic subtypes.39 This is intriguing in light of the fact that KRAS mutation appears to represent an early event in a similar fraction of colorectal carcinomas.40 Therefore, the translational component of this study will quantify expression of VEGF ligand, receptor, and activated receptor as well as EGF ligand, receptor, and activated receptor via immunohistochemistry in mucinous adenocarcinoma of the ovary to determine the potential utility of biologic agents directed at these proteins for inclusion in future studies of mucinous carcinomas of the ovary. We will also assess the mutational status of the KRAS oncogene in primary mucinous ovarian carcinomas obtained prior to treatment with oxaliplatin and capecitabine and then determine whether evidence exists for a correlation between the presence of KRAS mutation and response to therapy in this group of patients with ovarian cancer. Pathology Primary mucinous carcinomas of the ovary are uncommon, and historically it was reported that their biologic behavior and survival rates were better than that of serous carcinoma.41-43 This interpretation is complicated by several features not initially recognized, including the following: 1) more than 60% to 80% of mucinous carcinomas of the ovary are of Stage I;43-47 2) most mucinous carcinomas found in the ovary are metastatic rather than primary;42,47,48 3) primary mucinous carcinomas represent only 3-12% of mucinous ovarian neoplasms; 4) primary mucinous carcinomas represent fewer than 11% of ovarian carcinomas; and 5) the survival of advanced stage primary ovarian mucinous carcinoma appears to be significantly worse than that of serous carcinoma matched for stage.43-50 (M. Brady, personal communication) It is unclear whether the poor prognosis relates to an intrinsic aggressiveness of these tumors, a lack of sensitivity to chemotherapeutic agents effective in the therapy of serous carcinoma, or a failure to recognize some of the mucinous carcinomas as metastatic rather than primary to the ovary. In several recent reviews of mucinous tumors of the ovary, the authors have found that they had previously misdiagnosed a surprisingly large proportion of their cases as primary and later identified an occult gastric, colonic, pancreatic or other site of origin.47,48,51,52 These studies led to efforts to better delineate criteria which would distinguish primary from metastatic mucinous carcinomas of the ovary. Immunohistochemical stains of potential diagnostic utility include CK7, CK 20, CDX2, beta-catenin, P504S, MUC2 and MUC5AC.47,48,53,54 In addition, a variety of gross morphologic and histologic criteria have also been proposed in the last 3 years that appear to help in distinguishing primary from metastatic mucinous carcinomas of the ovary, including the following: features favoring a metastatic mucinous tumor - bilaterality, size less than 10cm, solid growth pattern, multinodularity, surface involvement by neoplastic cells, infiltrative pattern of stromal invasion, signet ring cells, hilar involvement, single cell invasion; features favoring a primary mucinous tumor - unilaterality, size greater than 10 cm, expansile -6- GOG-0241 pattern of invasion; complex papillary growth pattern.48 An algorithm based on a few features has been recently proposed that classified correctly 90% of ovarian mucinous carcinomas as primary or metastatic, as follows: all bilateral tumors, and unilateral tumors less than 10cm in diameter are metastatic, while those that are unilateral and greater than 10 cm are primary.52 In contrast to past experience, the vast majority of metastatic mucinous carcinomas of the ovary can now be reliably discriminated from those which arise in the ovary by routine histologic assessment.48,55 Previous phase III trials, principally from the GOG, have shown that mucinous histology is a poor prognostic factor. Taxane/platinum front-line chemotherapy appears to be relatively ineffective based on response rates and overall survival data. Based on colorectal cancer trials using regimens such as FOLFOX or XELOX, we hypothesize that the XELOX regimen will offer a better response, longer progression-free survival, and longer overall survival than current taxane/platinum strategies in patients with mucinous ovarian cancers. Almost concomitantly, UK investigators have independently developed an identical therapeutic strategy for this subset of epithelial ovarian cancer patients. An international collaboration as proposed is ideal for exploring this approach. While improvement in overall survival is the primary objective of this study, symptom assessment for neurotoxicity will be an important component of this trial. If this trial is positive, it would most certainly change the standard primary chemotherapy approach for women with mucinous cancers of the ovary. If negative, then other novel regimens should be pursued. 2.1 Inclusion of Women and Minorities The Gynecologic Oncology Group and GOG participating institutions will not exclude potential subjects from participating in this or any study solely based on ethnic origin or socioeconomic status. Every attempt will be made to enter all eligible patients into this protocol and therefore address the study objectives in a patient population representative of the entire ovarian carcinoma population treated by participating institutions. -4- 3.0 GOG-0241 PATIENT ELIGIBILITY AND EXCLUSIONS 3.1 Eligible Patients 3.11 Patients with a histologic diagnosis of mucinous adenocarcinoma of the ovary with either optimal (≤ 1 cm residual disease) or suboptimal residual disease following initial surgery (See Section 7.2). 3.12 All patients must have had appropriate surgery including appendectomy for ovarian or fallopian tube carcinoma with appropriate tissue available for histologic evaluation to confirm diagnosis and stage. 3.13 Patients must have stage II-IV disease or recurrent Stage I disease (chemonaïve). 3.14 Patients must be entered within 8 weeks of primary debulking surgery for their mucinous adenocarcinoma. 3.15 Patients with a negative colonoscopy within 1 year of enrolling in the study. 3.16 Patients must have adequate: 3.141 Bone marrow function: Absolute neutrophil count (ANC) ≥1,500/mcl, equivalent to Common Terminology Criteria for Adverse Events version 3.0 (CTCAE v3.0) grade 1. Platelets ≥100,000/mcl. (CTCAE v3.0 grade 0-1). Renal function: Creatinine less than or equal to 1.5 x institutional upper limit normal (ULN), CTCAE v3.0 grade 1. 3.142 Hepatic function: Bilirubin ≤1.5 x ULN (CTCAE v3.0 grade 1). SGOT and alkaline phosphatase ≤to 2.5 x ULN (CTCAE v3.0 grade 1). 3.143 Neurologic function: Neuropathy (sensory and motor) less than or equal to CTCAE v3.0 grade 1. 3.17 Urine dipstick for proteinuria <2+. If urine dipstick is 2+, 24 hour urine must demonstrate ≤ 1g protein in 24 hours. 3.18 Adequate coagulation parameters (within 14 days of randomization): 3.18.1 International Normalized Ratio (INR) ≤ 1.5xULN 3.18.2 Activated Pro Thrombin Time (APTT) ≤ 1.5xULN 3.19 Patients who have met the pre-entry requirements specified in Section 7.0. -5- 3.2 GOG-0241 3.20 Patients must have signed an approved informed consent and authorization permitting release of personal health information. 3.21 Patients with GOG Performance Grade of 0, 1 or 2. 3.22 Patients must be greater than or equal to 18 years of age. 3.23 Patients with life expectancy >3 months Ineligible Patients 3.21 Patients with known colon cancer or history of colon cancer. 3.22 Patients with primary peritoneal carcinoma. 3.23 Patients with a history of other invasive malignancies, with the exception of non-melanoma skin cancer, are excluded if there is any evidence of other malignancy being present within the last 5 years. Patients are also excluded if their previous cancer treatment contraindicates this protocol therapy. 3.24 Patients who have received either chemotherapy or radiotherapy for ovarian cancer prior to enrollment. 3.25 Patients with a major surgical procedure anticipated during the course of the study. 3.26 Patients with minor surgical procedures anticipated during the course of study. 3.261 Minor surgical procedures (i.e., mediport insertion), fine needle aspiration or core biopsies within 7 days prior to the first date of bevacizumab therapy. 3.27 Patients with major surgery or significant traumatic injury within 28 days prior to study entry. 3.28 Patients with a history of abdominal fistula or perforation within the past 12 months. 3.29 Patients with a current, serious, non-healing wound, ulcer, or bone fracture. 3.30 Patients who have had prior therapy with docetaxel or gemcitabine or bevacizumab. 3.31 Patients with known know hypersensitivity to Chinese hamster cell products or other recombinant human or humanized antibodies. 3.32 Patients with mixed epithelial ovarian cancer histology. 3.33 Patients with tumors of low malignant potential -6- GOG-0241 3.34 Patients with a history or evidence of upon physical examination of CNS disease, including history of primary brain tumor, or any history of brain metastases, or seizures not controlled with standard medical therapy. 3.35 Clinically significant cardiac function. Specifically, patient may not have: 3.351 Uncontrolled hypertension, defined as systolic > 150mm Hg or diastolic > 100mm Hg.Patients with a history of hypertension are permitted, but patient must have BP less than or equal to 140/90 mm Hg. Use of blood pressure medications to achieve and maintain blood pressure control is permitted. 3.352 Myocardial infarction or unstable angina within 6 months of the first date of bevacizumab therapy. 3.353 New York Heart Association (NYHA) Grade II or greater congestive heart failure (See Appendix II) or serious cardiac arrhythmia requiring medication. Women who have received prior treatment with anthracycline (including doxorubicin and/or liposomal doxorubicin) and have an ejection fraction < 50% will be excluded from the study. 3.354 Grade 1, Category 2 or greater, peripheral vascular disease (Please see Appendix III). Patient cannot have anything worse than mild, symptomatic claudication with exercise. 3.355 History of cerebrovascular accident (CVA, stroke), transient ischemic attack (TIA) or subarachnoid hemorrhage within six months of the first date of bevacizumab therapy. 3.36 History of pulmonary embolism or deep vein thrombosis in the past 6 months. 3.37 Patients with any symptoms or history of peripheral neuropathy. 3.38 Previous history of malabsorption or other conditions preventing oral treatment. 3.39 Patients who are pregnant or nursing. -74.0 GOG-0241 STUDY MODALITIES 4.1 Oxaliplatin (Eloxatin) (NSC #266046) 4.11 Other Names: l-OHP, SR96669 4.12 Chemical Name: trans-l-diaminocyclohexane oxalatoplatinum, cis[oxalato(trans-l-1,2-diaminocyclohexane) platinum (II)] 4.13 Molecular Formula: C8H14N2O4Pt M.W: 397.33 4.14 Description: Oxaliplatin is an organoplatinum complex in which the platinum atom is complexed with 1,2-diaminocyclohexane (DACH) with an oxalate ligand as a leaving group. Platinum content is 48.1% to 50.1%. White crystalline powder. 4.15 How Supplied: Freeze-dried powder for IV infusion in vials containing 50 mg and 100 mg of oxaliplatin. The excipient is lactose monohydrate, 450 mg and 900 mg respectively. The freeze-dried powder is stable for three years at room temperature. The pH of an aqueous solution of 2 mg/mL is between 4.8 and 5.7. 4.16 Solution Preparation: The powder is reconstituted by adding 10 mL (for the 50 mg vials) or 20 mL (for the 100 mg vials) of Water for Injection or Dextrose 5% in Water, which yields a 5 mg/mL solution. Do not administer the reconstituted solution without further dilution. This solution is then further diluted in an infusion solution of 250 mL to 500 mL Dextrose 5% in Water. The reconstitution or final dilution must never be performed with a sodium chloride solution. 4.17 Route of Administration: intravenous. (See Section 5.2) 4.171 Maximum body surface area used for dose calculations will be 2.0 m2 as per GOG Chemotherapy Procedures Manual. 4.18 Storage: Store at room temperature, protect from light. Shelf life: 3 years under these conditions. 4.19 Stability: Use product within 8 hours after withdrawing desired dose, as the product contains no bacteriostatic agent. Reconstituted solution: in Sterile Water for Injection or Dextrose 5% in Water in the original vial; the solution may be stored for 24 to 48 hours at 2 to 8 C. Infusion solution: after dilution in Dextrose 5% in Water, the shelf life is 24 hours at room temperature. -8- GOG-0241 4.110 Incompatibilities: Do not mix or administer with saline or other chloride-containing solution. Oxaliplatin is unstable in the presence of chloride. Do not combine with alkaline solutions such as 5-fluorouracil (pH 9.2) or other alkaline medications (pH >7) or media. Oxaliplatin is unstable under alkaline conditions and will degrade. Do not simultaneously administer other drugs by the same infusion line. All IV lines/catheters must be flushed with Dextrose 5% in Water, both before and after oxaliplatin administration. Do not use needles or intravenous infusion sets containing aluminum components for preparation or administration due to the risk of degradation of oxaliplatin upon contact with aluminum. 4.111 Supplier: Sanofi-synthelabo 4.112 Adverse Effects: In company studies, the WHO grading scale was used for the toxicities described below except for neurotoxicity. Sanofi developed a grading scale for the sensory neuropathies to accommodate the unique neuropathies observed (see below) 4.1121 Neurotoxicity: The most commonly observed oxaliplatin toxicity is acute and cumulative neurotoxicity, observed in patients treated at doses above 100 mg/m2/cycle. This neurotoxicity has included paresthesias and dysesthesias of the hands, feet, and perioral region as well as unusual pharyngo-laryngodysesthesias characterized by a loss of sensation of breathing without any objective evidence of respiratory distress (hypoxia, laryngospasm, or bronchospasm). Oxaliplatin neurotoxicity appears to be exacerbated by exposure to cold. Patients on this study will be counseled to avoid cold drinks and exposure to cold water or air. Should a patient develop pharyngo-laryngodysesthesia, their oxygen saturation should be evaluated via a pulse oximeter; if normal, an anxiolytic agent should be given and the patient observed in the clinic until the episode has resolved. Because this syndrome may be associated with the rapidity of oxaliplatin infusion, subsequent doses of oxaliplatin should be administered as a 6-hour infusion (instead of the normal 2hour infusion). Due to the unique and cumulative nature of the reversible neurotoxicity reported for oxaliplatin, Sanofi developed a grading scale for the sensory neuropathies, which has been used in all company-sponsored studies. A fusion of the NCI Common Toxicity Criteria (CTC) version 2.0 sensory neuropathy grading scale and the neurologic toxicity grading scale used -9- GOG-0241 in the company-sponsored studies is provided below (Table 4.1). This grading scale will be used for the evaluation of oxaliplatin-associated sensory neuropathies and for the determination of dose modifications. (For more information on dose modifications due to toxicity, see Section 6.) Table 4.1: Toxicity Scale for the Sensory Neuropathies Associated with Oxaliplatin Grade Symptoms Grade 0 None Grade 1 Paresthesias/dysesthesias* of short duration that resolve and do not interfere with function Grade 2 Paresthesias/dysesthesias* interfering with function, but not activities of daily living (ADL) Grade 3 Paresthesias/dysesthesias* with pain or with functional impairment that also interfere with ADL Grade 4 Persistent paresthesias/dysesthesias* that are disabling or life-threatening *May be cold-induced. Acute and cumulative neurotoxicities are dose limiting for oxaliplatin. The acute neurotoxicity is characterized by paresthesias and dysesthesias that may be triggered or exacerbated by exposure to cold. These symptoms occur within hours of exposure and are usually reversible over the following hours or days. Cumulative doses of oxaliplatin above 680 mg/m2 may produce functional impairment characterized by difficulty performing activities requiring fine sensory-motor coordination; impairment is caused by sensory rather than motor changes. The likelihood of experiencing neurotoxicity is directly related to the total cumulative dose of oxaliplatin administered. The relative risk of developing neurotoxicity was 10%, 50%, and 75% in patients who received total cumulative oxaliplatin doses of 780 mg/m2, 1,170 mg/m2, and 1,560 mg/m2, respectively. Both acute and cumulative neurotoxicities due to oxaliplatin have lessened in 82% of patients within 4 to 6 months, and have completely disappeared by 6 to 8 months in 41% of patients. In addition, the likelihood that neurologic symptoms will regress has been shown to correlate inversely with cumulative dose. Consider using prophylactic calcium gluconate and magnesium sulfate (administered in the same bag), 1 g each, delivered i.v. over 15 min just before the oxaliplatin infusion and repeated at the same dose after the completion of the oxaliplatin infusion. Ca gluconate and Mg sulfate were -10- GOG-0241 given. Ca/Mg infusions should not be administered to patients with known hypercalcemia, or treated with thiazidie diuretics or digitalis.40 Clinical ototoxicity occurs in less than 1% of patients following oxaliplatin administration, and severe ototoxicity has not been reported. Rarely, mental status changes and confusion have been associated with oxaliplatin in combination with 5-FU/LV. In one patient with a grade 5 infection and confusion after treatment with oxaliplatin and 5-FU/LV, work-up revealed a cerebrovascular accident. A seizure has been reported with the treatment of oxaliplatin. 4.1122 Hematologic: Single-agent oxaliplatin generally produces only mild hematologic toxicity; no Grade 4 anemia has been reported, and less than 1% of patients have Grade 4 neutropenia or thrombocytopenia. When oxaliplatin is combined with 5-FU/LV schedules for which myelosuppression is dose-limiting, the resulting toxicity is greater than that observed with the 5-FU/LV regimen alone. 4.1123 Gastrointestinal: Nausea and vomiting are common side effects of oxaliplatin treatment and require premedication with antiemetic medications (anti-5HT 3 medications are effective). Diarrhea is also common, and occurs in 44% of patients treated with single-agent oxaliplatin and 68% of those treated with oxaliplatin plus 5-FU/LV. The addition of oxaliplatin to the regimen produces a significant increase in both the frequency and severity of diarrhea and mucositis usually expected in patients receiving 5-FU/LV on an infusion schedule. The addition of oxaliplatin to 5-FU/LV has also been associated with ileus, enterocolitis, and typhilitis. Rarely, this has been associated with ischemic bowel requiring bowel resection. Grade 1-2 elevation of liver enzymes is common during oxaliplatin therapy. Elevation of liver enzymes is often associated with hepatic metastases. 4.1124 Cutaneous: Erythema or skin eruptions are uncommon with single agent oxaliplatin, and the incidence of alopecia is < 2%. The incidence of cutaneous toxicity is much higher in combination with 5-FU (overall 22%; Grade 3 & 4 - 2%). When oxaliplatin in combination with 5-FU is administered by infusion, the incidence of alopecia is higher (6% overall; Grade 3 & 4 - 2%). 4.1125 Allergic Reactions: Other platinum compounds are associated with allergic reactions, but such reactions have been uncommon with oxaliplatin and have varied from rashes to anaphylaxis. Severe allergic reactions were reported in 0.5% of patients during clinical development. One patient has died of an anaphylactic-like reaction. In all other known cases, the reaction resolved with symptomatic treatment. 4.1126 Pulmonary: Pulmonary fibrosis and adult respiratory distress syndrome (ARDS) are rarely associated with this agent. During the period from 1996 to April 2000, approximately 9,000 patients were treated in clinical trials and 45,000 patients were treated with oxaliplatin in post-marketing -11- GOG-0241 and compassionate use studies. During this period, pulmonary fibrosis was suspected in eleven patients. On external review, a relationship with oxaliplatin was thought to be possible in at least six of the eleven patients. Among the eleven patients, four patients died, two of which were due to pulmonary fibrosis. In addition, two cases of eosinophilic pneumonia and one case of interstitial pneumonitis were reported for which the causal relationship with oxaliplatin could not be excluded. 4.1127 Other: Other toxicities include fever (with or without infection) in 15% of patients receiving monotherapy and 5% of those receiving oxaliplatin in combination therapy. Transient decreased vision has been reported in < 0.1% of patients. Rarely has significant cardiac toxicity with death been observed in patients treated with oxaliplatin. Three patients experienced reversible supraventricular arrhythmias during oxaliplatin administration; one of these patients tolerated additional cycles of oxaliplatin with a recurrence of the arrhythmia. (Hypotension and syncope have been observed with oxaliplatin treatment). Few cases of hemolytic-uremic syndrome and disseminated intravascular coagulation (DIC), phlebitis, and extravasations have been observed. Oxaliplatin monotherapy is associated with an extremely low incidence of treatment-related deaths (1 of 244 patients; 0.4%). The single patient who died of an acute anaphylactic reaction following oxaliplatin was receiving ondansetron concurrently. One patient with pre-existing pulmonary fibrosis had continued worsening of the condition, subsequently developed sepsis, and died; the investigator thought the relationship to oxaliplatin was unknown. The incidence of treatment related deaths following combination 5-FU/oxaliplatin therapy was 1.1% (13 of 1183 patients), similar to the incidence seen with 5-FU alone (Sanofi Clinical Investigator's Brochure, May 1998). 4.2 Capecitabine (Xeloda) (NSC #712807) 4.21 Formulation: Capecitabine is supplied as biconvex, oblong, film-coated tablets. 150 mg tablets are light peach in color engraved with Xeloda on one side and 150 on the other and come packaged in bottles of 120. 500 mg tablets are peach in color engraved with Xeloda on one side and 500 on the other and come packaged in bottles of 240. 4.22 Storage: Store at room temperature 25C (77F), excursions permitted to 1530C (59-89F). Keep tightly closed. 4.23 Administration: The patient will take a daily dose of 2000 mg/m2 in two divided doses for 14 consecutive days followed by seven-day rest period. The tablets are to be swallowed with water and taken within 30 minutes after the end of a meal. (See Sec. 5.22) 4.24 Adverse effects: Hematologic: neutropenia, thrombocytopenia, anemia, lymphopenia Gastrointestinal: diarrhea, nausea, vomiting, stomatitis, abdominal pain, constipation, dyspepsia, hyperbilirubinemia, anorexia, dehydration -12- GOG-0241 Skin: hand and foot syndrome, dermatitis, edema Neurological: paresthesias, headache, dizziness, insomnia Constitutional: fatigue, fever, myalgias Ophthalmic: eye irritation Alteration of coagulation parameters in patients taking anticoagulants was also seen. 4.25 4.3 Supplier: Commercially available. Roche laboratories In the event that a patient cannot obtain study drug (indigent patient, no insurance, refusal of third party payer), contact Roche Patient Assistance Program: 1-800-443-6676, selection #2. Carboplatin (Paraplatin, NSC #241240) 4.31 Formulation: Carboplatin is supplied as a sterile lyophilized powder available in single-dose vial containing 50 mg, 150 mg and 450 mg of carboplatin for administration by intravenous infusion. Each vial contains equal parts by weight of carboplatin and mannitol. 4.32 Preparation: Immediately before use, the content of each vial must be reconstituted with either sterile water for injection, USP, 5% dextrose in water, or 0.9% sodium chloride injection, USP, according to the following schedule: Vial strength Diluent volume 50 mg 150 mg 450 mg 5 ml 15 ml 45 ml These dilutions all produce a carboplatin concentration of 10 mg/ml. When prepared as directed, carboplatin solutions are stable for eight hours at room temperature, since no antibacterial preservative is contained in the formulation it is recommended that carboplatin solutions be discarded eight hours after dilution. NOTE: Aluminum reacts with carboplatin causing precipitate formation and loss of potency, therefore, needles or intravenous sets containing aluminum parts that may come in contact with the drug must not be used for the preparation or administration of carboplatin. 4.33 Storage: Unopened vials of carboplatin are stable for the life indicated on the package when stored at controlled room temperature and protected from light. 4.34 Adverse effects: Some of the adverse events expected with carboplatin treatment are listed below. Hematologic: Myelosuppression is the major dose-limiting toxicity. Thrombocytopenia, neutropenia, leukopenia, and anemia are common, but typically resolve by Day 28 when carboplatin is given as a single agent. -13- GOG-0241 Allergic reactions: Hypersensitivity to carboplatin has been reported in 2% of patients receiving the drug as first line therapy and in approximately 28% of patients being retreated with Carboplatin after receiving platinum as first line therapy. Symptoms include rash, urticaria, erythema, pruritus, and rarely bronchospasm and hypotension. The reactions can be successfully managed with standard epinephrine, corticosteroid, and antihistamine therapy. Neurologic: Peripheral neuropathies have been observed in 4% of patients receiving carboplatin with mild paresthesia being the most common. Gastrointestinal: Nausea and vomiting are the most common GI events; both usually resolve within 24 hours and respond to antiemetics. Other GI events include diarrhea, weight loss, constipation, and gastrointestinal pain. Hepatic toxicity: Elevated alkaline phosphatase, total bilirubin, and SGOT have been observed. Metabolic: Hypomagnesemia, hypokalemia, hypocalcemia. Secondary malignancy: Potential risk of other malignancies when used in multi-agent therapy. Renal toxicity: Increased serum creatinine and blood urea nitrogen are uncommon, mild, and usually reversible. Other: Pain and asthenia are the most common miscellaneous adverse events. Alopecia has been reported in 3% of the patients taking carboplatin. 4.35 Supplier: Commercially available. Bristol-Myers Squibb Co.* 4.36 Administration: Carboplatin dosed to an AUC 5 mg/ml/min will be given IV every 3 weeks. Pretreatment with a 5-HT3 inhibitor and dexamethasone 20 mg IV or PO 30 minutes prior to infusion is suggested. The carboplatin infusion should begin after the Cetuximab infusion and may begin during the observation period. *Refer to package insert for additional information. 4.4 Paclitaxel (NSC #673089) 4.41 Formulation: Paclitaxel is supplied as a 6mg/mL non-aqueous solution in multidose vials containing 30mg/5mL, 100mg/16.7mL, or 300mg/50mL of paclitaxel. In addition to 6mg of paclitaxel, each mL of sterile non-pyrogenic solution contains 527mg of purified Cremophor® EL (polyoxyethylated castor oil) and 49.7% (v/v) dehydrated alcohol, USP. 4.42 Storage: Unopened vials of paclitaxel are stable to the date indicated on the package when stored between 20 to 25°C (68 to 77°F). Protect from light. 4.43 Preparation: Paclitaxel must be diluted prior to infusion. Paclitaxel should be diluted in 0.9% Sodium Chloride for Injection, USP; 5% Dextrose Injection, USP; 5% Dextrose and 0.9% Sodium Chloride Injection, USP; or 5% Dextrose in Ringer’s Injection to a final concentration of 0.3 to 1.2mg/mL. The solutions -14- GOG-0241 are physically and chemically stable for up to 27 hours at ambient temperature (approximately 25°C / 77°F) and room lighting conditions. NOTE: In order to minimize patient exposure to the plasticizer DEHP, which may be leached from PVC infusion bags or sets, diluted paclitaxel solutions should be stored in bottles (glass, polypropylene) or plastic (polypropylene, polyolefin) bags and administered through polyethylene-lined administration sets. Paclitaxel should be administered through an inline filter with a microporous membrane not greater than 0.22 microns. Use of filter devices such as IVEX-2® or IVEX-HP®, which incorporate short inlet and outlet PVC-coated tubing, has not resulted in significant leaching of DEHP. All patients should be premedicated with corticosteroids, diphenhydramine, and H2 antagonists prior to paclitaxel administration in order to prevent severe hypersensitivity reactions. Patients who experience severe hypersensitivity reactions to paclitaxel should not be rechallenged with the drug. Adverse Effects: Consult the package insert for the most current and complete information. Incidence rates of adverse events associated with paclitaxel are provided in the product package insert. The following events are expected with the administration of paclitaxel: Hematologic: Myelosuppression is the major dose-limiting toxicity. Neutropenia is both dose-and schedule-dependent and typically resolves rapidly. Fever is common and infectious episodes are seen in about 1/3 of the patients receiving paclitaxel. Thrombocytopenia is uncommon and the cases that occur are usually mild to moderate. Bleeding episodes may occur. While anemia is common, it is severe only in 16% of the cases. Allergic reactions: Although patients are premedicated, hypersensitivity reactions still occur in approximately 40% of patients receiving paclitaxel (20% of cycles). Severe reactions are rare and generally occur within the first hour of administration; no severe reactions have been reported after the third cycle. The most common symptoms observed in severe reactions include dyspnea, flushing, chest pain, and tachycardia. Minor hypersensitivity reactions include flushing, rash, hypotension, dyspnea, tachycardia, and hypertension. Cardiovascular: Cardiovascular events observed with paclitaxel therapy include hypotension and bradycardia; typically, neither discontinuation of paclitaxel nor specific therapy for the event is required. Cardiovascular events that are possibly related to paclitaxel therapy occur in approximately 1% of patients and include syncope, rhythm abnormalities (asymptomatic ventricular tachycardia, bigeminy, and complete AV block requiring a pacemaker), hypertension, and venous thrombosis. Neurologic: The frequency and severity of neurologic events are dosedependent. Peripheral neuropathy is rarely severe and may be the cause of paclitaxel discontinuation in 1% of patients. Sensory symptoms usually improve or resolve within several months of paclitaxel discontinuation. Serious neurologic events, such as grand mal seizures, syncope, ataxia, and neuroencephalopathy, are rare. -15- GOG-0241 Gastrointestinal: The most common GI toxicities, which include nausea, vomiting, diarrhea, and mucositis, are typically mild or moderate and rarely severity. Mucositis is schedule-dependent and occurs more frequently with a 24-hour infusion than a 3-hour infusion of paclitaxel. Other: Although 60% of all patients experience arthralgia and myalgia, there is no consistent relationship between the dose or schedule of paclitaxel and the frequency of these events. The symptoms, which usually begin 2 or 3 days after paclitaxel treatment, are generally transient. Injection site reactions are more common with the 24-hour infusion of paclitaxel and are typically mild, consisting of erythema, tenderness, skin discoloration, or swelling at the injection site. Almost all of the patients receiving paclitaxel experience alopecia, but nail changes are uncommon. Edema may occur, but it is rarely severe enough to lead to discontinuation of treatment. Paclitaxel Premedication 4.44 Patients should receive decadron 20 mg p.o. 14 and 7 hours prior to paclitaxel administration. Intravenous premedication as listed, or institutional guidelines may be followed. 4.44 4.5 Supplier: Commercially available from both Bristol-Myers Squibb Oncology as well as generic manufacturers. Consult the American Hospital Formulary Service Drug Information guide, Facts and Comparisons, or the package insert for additional information. Bevacizumab (NSC #704865, IND #7921) 4.51 Other Name: rhuMAb VEGF 4.52 Molecular Weight: Approximate molecular weight is 149,000 daltons 4.53 Description: Bevacizumab is a recombinant humanized anti-VEGF monoclonal antibody, consisting of 93% human and 7% murine amino acid sequences. The agent is composed of human IgG framework and murine antigen-binding complementarity-determining regions. Bevacizumab blocks the binding of vascular endothelial growth factor (VEGF) to its receptors resulting in inhibition of angiogenesis. 4.54 How supplied: Bevacizumab is a clear to slightly opalescent, colorless to pale brown, sterile liquid concentrate for solution for IV infusion. Bevacizumab will be supplied in 5-mL (100 mg) or 20-mL (400 mg) glass vials containing 4-mL or 16-mL of bevacizumab, respectively (25 mg/mL for either vial). Vials contain bevacizumab with phosphate, trehalose, polysorbate 20, and Sterile Water for Injection (SWFI), USP. Vials contain no preservative and are suitable for single use only. 4.55 Storage and Stability: Upon receipt of bevacizumab, vials are to be refrigerated at 2C-8C (36F-46F) and should remain refrigerated until just prior to use. DO NOT FREEZE. DO NOT SHAKE. PROTECT FROM LIGHT. Open vials -16- GOG-0241 must be used within 8 hours. VIALS ARE FOR SINGLE USE ONLY. Vials used for one subject may not be used for any other subject. 4.56 Preparation: Vials contain no preservative and are intended for single use only. Place the calculated dose in 100mL of 0.9% sodium chloride for injection. Once diluted in 0.9% Sodium Chloride for Injection, the bevacizumab solution must be administered within 8 hours. 4.57 Administration: Bevacizumab is administered intravenously as a continuous infusion. The dose should be based on the patient’s actual body weight; the dose will be recalculated if there is a weight change of > 10% from baseline. The initial dose should be administered over a minimum of 90 minutes. If no adverse reactions occur after the initial dose, the second dose should be administered over a minimum of 60 minutes. If no adverse reactions occur after the second dose, all subsequent doses should be administered over a minimum of 30 minutes. If infusion-related adverse reactions occur at any time, patient should be pre-medicated (see section 5.2) prior to next dose and infusion time may not be reduced for the subsequent infusion. To insure complete delivery of bevacizumab, the IV infusion line must be flushed with 0.9% sodium chloride. The following are two recommended methods for flushing the bevacizumab IV infusion line: 1. 2. When the bevacizumab infusion is complete, add an additional 50mL of 0.9% sodium chloride for injection to the bevacizumab infusion bag. Continue the infusion until a volume equal to that of the volume contained in the tubing has been administered. Replace the empty bevacizumab infusion bag with a 50mL bag of 0.9% sodium chloride for injection and infuse a volume equal to the volume contained in the tubing. Please note: The flush is not included in the total recommended infusion times. 4.58 Clinical Supplies: Bevacizumab (NSC 704865) will be provided free of charge by Genentech and distributed by the Pharmaceutical Management Branch (PMB), Cancer Therapy Evaluation Program (CTEP), Division of Cancer Treatment and Diagnosis (DCTD), National Cancer Institute (NCI). 4.59 Drug Ordering and Accountability NCI supplied agents may be requested by the Principal Investigator or their authorized designee at each participating institution. Pharmaceutical Management Branch (PMB) policy requires that drug be shipped directly to the institution where the patient is being treated. PMB does not permit the transfer of agents between institutions (unless prior approval from PMB is obtained). The CTEP assigned protocol number must be used for ordering all CTEPsupplied investigational agents. The responsible investigator at each participating institution must be registered with CTEP, DCTD, through an annual submission of FDA Form 1572, Supplemental Investigator Data Form (IDF), and Financial Disclosure Form. If there are several participating investigators at one institution, CTEP supplied investigational agents for the -17- GOG-0241 study should be ordered under the name of one lead investigator at that institution. Agent may be requested by completing a Clinical Drug Request (CDR [NIH986]) available on the CTEP web site at http://ctep.cancer.gov/requisition/ and faxing it to the Pharmaceutical Management Branch, DCTD, NCI, EPN, Room 7149, Bethesda, MD 20892 or faxing it to (301) 480-4612. Beginning 11/10/03, all CDR’s for PMB-distributed agents sent to the PMB must be signed by the investigator in whose name the agent is ordered OR by the shipping designee OR by an ordering designee whom the investigator has listed on their most recent IDF on file with the PMB. If the investigator has not designated the individual signing the CDR as a shipping or ordering designee, or if the shipping or ordering designees at a clinical site change, the first two pages of the IDF should be updated to reflect the current designees, the IDF should be signed and dated by the investigator and returned to the PMB by fax at (301) 402-4870. For questions (or the first two pages of the investigator’s current IDF) call the PMB at (301) 496-5725 Monday through Friday from 8:30 am to 4:30 pm Eastern Time. The Investigator, or a responsible party designated by the investigator, must maintain a careful record of the inventory and disposition of all agents received from the PMB using the NCI Drug Accountability Record Form (DARF). See the CTEP web site for Policy and Guidelines for Accountability and Storage of Investigational Drugs at http://ctep.cancer.gov/requisition/ Requests for Investigator’s Brochures (IB) should be e-mailed to ibcoordinator@mail.nih.gov or you may call the IB coordinator at 301-4965725. 4.591 Drug Returns: Only unreconstituted drug supplies should be returned to the PMB. When it is necessary to return study drug, investigators should return the study drug to the PMB using the NCI Return Drug List available on the CTEP home page (http://ctep.cancer.gov) or by calling the PMB at 301-4965725. 4.592 Comprehensive Adverse Events and Potential Risks List (CAEPR) for Bevacizumab (NSC #704865) The Comprehensive Adverse Event and Potential Risks list (CAEPR) provides a single, list of reported and/or potential adverse events (AE) associated with an agent using a uniform presentation of events by body system. In addition to the comprehensive list, a subset, the Agent Specific Adverse Event List (ASAEL), appears in a separate column and is identified with bold and italicized text. This subset of AEs (ASAEL) contains events that are considered 'expected' for expedited reporting purposes only. Refer to the 'CTEP, NCI Guidelines: Adverse Event Reporting Requirements' http://ctep.cancer.gov/reporting/adeers.html for further clarification. The CAEPR does not provide frequency data; refer to the Investigator's Brochure for this information. Below is the CAEPR for Bevacizumab. -18- GOG-0241 Version 1.2, June 19, 20071 Category Adverse Events with Possible (Body Relationship to Bevacizumab System) (CTCAE v3.0 Term) ALLERGY/IMMUNOLOGY Allergic reaction/hypersensitivity (including drug fever) Allergic rhinitis (including sneezing, nasal stuffiness, postnasal drip) 'Agent Specific Adverse Event List' (ASAEL) Allergic reaction/hypersensitivity (including drug fever) Allergic rhinitis (including sneezing, nasal stuffiness, postnasal drip) BLOOD/BONE MARROW Hemoglobin Leukocytes (total WBC) Neutrophils/granulocytes (ANC/AGC) Hemoglobin Leukocytes (total WBC) Neutrophils/granulocytes (ANC/AGC) CARDIAC ARRHYTHMIA Supraventricular arrhythmia NOS Ventricular fibrillation Supraventricular arrhythmia NOS CARDIAC GENERAL Cardiac ischemia/infarction Cardiac troponin I (cTnI) Hypertension Hypotension Left ventricular diastolic dysfunction Left ventricular systolic dysfunction Cardiac ischemia/infarction Hypertension CONSTITUTIONAL SYMPTOMS Fatigue (asthenia, lethargy, malaise) Fever (in the absence of neutropenia, where neutropenia is defined as ANC <1.0 x 109/L) Rigors/chills Weight loss Fatigue (asthenia, lethargy, malaise) Fever (in the absence of neutropenia, where neutropenia is defined as ANC <1.0 x 109/L) Rigors/chills DERMATOLOGY/SKIN Pruritus/itching Rash/desquamation Ulceration Urticaria (hives, welts, wheals) Wound complication, non-infectious Pruritus/itching Rash/desquamation Urticaria (hives, welts, wheals) GASTROINTESTINAL Anorexia Colitis Constipation Diarrhea Fistula, GI - Select Heartburn/dyspepsia Ileus (functional obstruction of bowel, i.e., neuroconstipation) Leak (including anastomotic), GI: large bowel Mucositis/stomatitis (functional/symptomatic) - Select Nausea Perforation, GI - Select Ulcer, GI - Select Vomiting Anorexia Constipation Diarrhea Heartburn/dyspepsia Mucositis/stomatitis (functional/symptomatic) - Select Nausea Vomiting HEMORRHAGE/BLEEDING Hemorrhage, GI - Select Hemorrhage, CNS Hemorrhage, GU: vagina Hemorrhage, pulmonary/upper respiratory: lung Hemorrhage, pulmonary/upper respiratory: nose Hemorrhage GI - Select Hemorrhage, CNS Hemorrhage, GU: vagina Hemorrhage, pulmonary/upper respiratory: lung Hemorrhage, pulmonary/upper respiratory: nose -19- Category (Body System) INFECTION Adverse Events with Possible Relationship to Bevacizumab (CTCAE v3.0 Term) GOG-0241 'Agent Specific Adverse Event List' (ASAEL) Infection with normal ANC or Grade 1 or 2 neutrophils Select Infection with normal ANC or Grade 1 or 2 neutrophils Select (pelvis, peritoneal cavity, rectum, scrotum, skin, wound) METABOLIC/LABORATORY Alkaline phosphatase ALT, SGPT (serum glutamic pyruvic transaminase) AST, SGOT (serum glutamic oxaloacetic transaminase) Bilirubin (hyperbilirubinemia) Creatinine Proteinuria Alkaline phosphatase ALT, SGPT (serum glutamic pyruvic transaminase) AST, SGOT (serum glutamic oxaloacetic transaminase) Bilirubin (hyperbilirubinemia) Proteinuria NEUROLOGY CNS cerebrovascular ischemia Dizziness Neurology - Other: (Leukoencephalopathy syndrome including reversible posterior leukoencephalopathy syndrome [RPLS]) CNS cerebrovascular ischemia Dizziness Pain - abdomen NOS Pain - chest/thorax NOS Pain - head/headache Pain - joint Pain - muscle Pain - NOS Pain - abdomen NOS Pain - chest/thorax NOS Pain - head/headache Pain - joint PAIN PULMONARY/UPPER RESPIRATORY Bronchospasm, wheezing Cough Dyspnea (shortness of breath) Fistula, pulmonary/upper respiratory - Select Nasal cavity/paranasal sinus reactions Voice changes/dysarthria (e.g., hoarseness, loss or alteration in voice, laryngitis) Pulmonary/Upper Respiratory - Other (nasal-septal perforation) Cough Dyspnea (shortness of breath) Nasal cavity/paranasal sinus reactions Voice changes/dysarthria (e.g., hoarseness, loss or alteration in voice, laryngitis) RENAL/GENITOURINARY Fistula, GU - Select Renal failure SYNDROMES Cytokine release syndrome/acute infusion reaction Cytokine release syndrome/acute infusion reaction VASCULAR Thrombosis/thrombus/embolism Visceral arterial ischemia (non-myocardial) 1This Thrombosis/thrombus/embolism table will be updated as the toxicity profile of the agent is revised. Updates will be distributed to all Principal Investigators at the time of revision. The current version can be obtained by contacting ADEERSMD@tech-res.com. Your name, the name of the investigator, the protocol and the agent should be included in the e-mail. -20- GOG-0241 Also reported on bevacizumab trials but with the relationship to bevacizumab still undetermined: BLOOD/BONE MARROW - Idiopathic thrombocytopenia purpura; platelets CARDIAC GENERAL - Cardiac arrest; pericardial effusion; pulmonary hypertension COAGULATION - DIC DEATH - Sudden death (cause unknown) DERMATOLOGY/SKIN - Hypopigmentation GASTROINTESTINAL - Rectal abscess/necrosis; small bowel obstruction; taste alteration METABOLIC/LABORATORY - Hyperglycemia; hypoglycemia; hypomagnesemia; hyponatremia MUSCULOSKELETAL/SOFT TISSUE - Aseptic necrotic bone; gait/walking; myasthenia gravis NEUROLOGY - Aseptic meningitis; confusion; peripheral neuropathy; seizure; syncope OCULAR/VISUAL - Cataract; watery eye PULMONARY/UPPER RESPIRATORY - ARDS; pneumonitis/pulmonary infiltrates; pneumothorax RENAL/GENITOURINARY - Urinary frequency Note: Bevacizumab in combination with other agents could cause an exacerbation of any adverse event currently known to be caused by the other agent, or the combination may result in events never previously associated with either agent. -21- 5.0 GOG-0241 TREATMENT PLAN AND ENTRY/RANDOMIZATION PROCEDURE All initial and continuing reviews must be submitted to the CTSU Regulatory Office. A CTSU IRB/Regulatory Approval Transmittal Sheet should be submitted along with the CTSU IRB Certification Form or its equivalent. (CTSU forms can be downloaded at https://www.ctsu.org/rss2_page.asp). IRB submissions can be faxed or mailed to: CTSU Regulatory Office Coalition of National Cancer Cooperative Groups 1818 Market Street, Suite 1100 Philadelphia, PA 19103 1-888-823-5923 FAX 215-569-0206 5.1 Patient Entry and Registration When a suitable candidate has been obtained for protocol entry, the following steps should be taken: 5.11 An approved informed consent form and authorization permitting release of personal health information must be signed by the patient or guardian. Current FDA, NCI, and institutional regulations concerning informed consent will be followed. 5.12 All eligibility requirements indicated in section 3.0 have been satisfied. 5.13 Fast Fact Sheet data should be gathered. 5.14 The institution must register the patient using the web-based registration application or by phone if necessary (800-523-2917). Instructions for web-based registration and randomization can be found by going to the GOG Web Menu page, selecting “Start/finish a patient registration,” and then selecting “Directions” found on the left side of the page. 5.15 Entry/Randomization will take place on the telephone after consideration of Fast Fact Sheet data. 5.16 The institution will enter the patient’s name, GOG number, and assigned regimen in the appropriate place in their Log Book to verify the patient’s entry. 5.17 Treatment should start within 14 days of randomization and no later than 6 weeks after surgery (if applicable). For patients randomized to receive bevacizumab, treatment with bevacizumab should not start until at least 28 days after surgery due to concerns about its impact on wound healing. It is recognized that there may be clinical reasons to start chemotherapy within 28 days of surgery; therefore, for patients randomized to receive bevacizumab, there is the option to give the first cycle of chemotherapy without bevacizumab. In this situation, the missed dose of bevacizumab will not be replaced and patients will receive 5 doses of bevacizumab with chemotherapy and 12 cycles as -22- GOG-0241 maintenance. Investigators will be asked to state at randomization whether or not they intend to start chemotherapy before 4 weeks have elapsed after surgery. 5.2 Treatment Plan 5.21 Arm I – 6 cycles of: Paclitaxel Carboplatin 175 mg/m2 IV over 3 h on day 1, q 21 days, followed by AUC = 6 on day 1, q 21 days 5.211 Paclitaxel will be administered as a 3-hour infusion on this study. For all courses in which paclitaxel is to be administered, it is recommended that a preparative regimen be employed to reduce the risk associated with hypersensitivity reactions. This regimen should include dexamethasone (either IV or PO), anti-histamine H1 (such as diphenhydramine) and anti-histamine H2 (such as cimetidine, ranitidine, or famotidine). 5.212 The dose of carboplatin will be calculated to reach a target area under the curve (AUC) of concentration x time according to the Calvert formula using an estimated glomerular filtration rate (GFR) from the Jelliffe formula. The initial dose of carboplatin must be calculated using GFR. In the absence of new renal obstruction or other renal toxicity greater than or equal to CTC grade 2 (serum creatinine >1.5 x ULN), the dose of carboplatin will not be recalculated for subsequent cycles, but will be subject to dose modification as noted. In patients with abnormally low serum creatinine (less than or equal to 0.6 mg/dl), due to reduced protein intake and/or low muscle mass, the creatinine clearance should be estimated using a minimum value of 0.6 mg/dl. If a more appropriate baseline creatinine value is available within 4 weeks of treatment that may also be used for the initial estimation of GFR. Calvert Formula: Carboplatin dose (mg) = target AUC x (GFR + 25) For the purpose of this protocol, the GFR is considered to be equivalent to the creatinine clearance. The creatinine clearance (Ccr) is estimated by the method of Jelliffe using the following formula for females: GFR = 0.9 x (98 - 0.8 (age* - 20)) Cr (mg/dl) *Age rounded to the nearest decade Where Ccr Age Scr = = = estimated creatinine clearance in ml/min patient’s age in years (from 20-80) serum creatinine in mg/dl In the absence of new renal obstruction or elevation of serum creatinine above 1.5 x ULN (CTC grade 2), the dose of carboplatin will not be recalculated for subsequent cycles, but will be subject to dose modification for hematologic criteria and other events as noted. -23- GOG-0241 The conversion equation below should be used for converting the IDMS serum creatinine (SrCr) value to the non-IDMS serum creatinine (SrCr) value prior to using the Calvert Formula to calculate the carboplatin dose. Non-IDMS SrCr (mg/dL) = IDMS SrCr (mg/dL) x 1.065 +0.067 Conversion of Common SrCr Values: 5.22 Clinic Station Reported IDMS SrCr Value (mg/dL) Needs to be converted to Non-IDMS SrCr to use for Carboplatin dosing (mg/dL) Less than 0.6 0.7 0.8 0.9 1 1.1 1.2 1.3 1.5 Use assigned 0.8 0.8 0.9 1.0 1.1 1.2 1.3 1.5 1.7 Arm II– 6 cycles of: Oxaliplatin 130 mg/m2 IV infusion over 2 h, Day 1, q 21 days Capecitabine 850 mg/m2 orally twice daily, Days 1-14, followed by 7-day rest period 5.221 This is an outpatient regimen. All patients should be given antiemetics (e.g. ondansetron or granisetron and dexamethasone) prior to and after the oxaliplatin infusion. On day 1 of each 21-day treatment cycle, patients receive 130 mg/m² oxaliplatin diluted in 250 to 500 mL Dextrose 5% in Water infused intravenously over 2 hours through a peripheral or central vein. Following the infusion of oxaliplatin, the infusion line should be flushed immediately with Dextrose 5% in Water. Cycles will be repeated every 21 days. Precautions and Warnings Regarding Oxaliplatin Administration: -24- GOG-0241 Oxaliplatin is unstable in the presence of chloride or alkaline solutions. Do NOT mix or administer oxaliplatin with saline or other chloride-containing solutions. Do NOT administer other drugs or solutions in the same infusion line. Flush IV lines/catheters with Dextrose 5% in Water both before and after oxaliplatin administration. All patients must be premedicated for nausea and vomiting using the standard antiemetic regimen for platinum-based therapies at the institution. [Anti-5HT 3 medications are effective]. Patients on this study should be counseled to avoid cold drinks and exposure to cold water or air because the neurotoxicity often seen with oxaliplatin appears to be exacerbated by exposure to cold. The period of time during which the patient is at risk for these coldinduced sensory neuropathies is not well documented. Patients should exercise caution regarding cold exposure during the treatment period. Peripheral sensory neuropathies can occur at any time after receiving oxaliplatin therapy. 5.222 The patient will also take a daily dose of capecitabine 1700 mg/m2 (the total daily dose will be divided in two doses given approximately every 12 hours) starting on day 1 of each 21 day cycle for 14 consecutive days followed by 7-day rest period. The tablets are to be swallowed with water and taken within 30 minutes after the end of a meal. If a patient vomits or misses a dose, they must not take the missed dose at all and must not double the next dose. Instead, patients should continue with the dosing as scheduled. -25- GOG-0241 5.223 Antiemetic Regimens It is anticipated that nausea and vomiting may be a significant side effect of each regimen. The following representative antiemetic regimens are suggested: Ondansetron 8-32 mg IV 30 minutes prior to administration of chemotherapy and dexamethasone 10-20 mg IV 30 minutes prior to drug administration, or, Granisetron 10 mcg/kg IV (or 2 mg PO) 30 minutes prior to chemotherapy, with or without lorazepam 0.5-2.0 mg IV 30 minutes prior to chemotherapy. 5.224 The minimum treatment period will be one course. 5.225 If side effects are not severe, a patient may remain on the study agents until 6 cycles are completed or documented disease progression. 5.226 Maximum body surface area used for dose calculations will be 2.0 m2 as per GOG Chemotherapy Procedures Manual. (See Table 5.1 for calculation of capecitabine dose) Table 5.1: Calculation of Dose and Dose Administration for Capecitabine Number of tablets to be taken in the 100% Dose Level Morning = twice daily 850 mg/m2 Evening Surface Area (m2) Total Daily Dose (mg)* 150 mg 500 mg 150 mg 500 mg < 1.6 2300 1 2 1 2 < 1.6-1.8 3000 0 3 0 3 > 1.8 3300 1 3 1 3 Number of tablets to be taken in the 75% Dose Level Morning = twice daily 637.5 mg/m2 Evening Surface Area (m2) Total Daily Dose (mg)* 150 mg 500 mg 150 mg 500 mg < 1.6 1750 2 1 3 1 < 1.6-1.8 2300 1 2 1 2 > 1.8 2600 2 2 2 2 -26- GOG-0241 Number of tablets to be taken in the 50% Dose Level Morning = twice daily 425 mg/m2 Evening Surface Area (m2) Total Daily Dose (mg)* 150 mg 500 mg 150 mg 500 mg < 1.6 1150 0 1 1 1 < 1.6-1.8 1500 0 1 0 2 > 1.8 1600 2 1 2 1 5.23 Arm III--Bevacizumab When Given With Carboplatin + Paclitaxel Six cycles of chemotherapy (See Section 5.21) plus bevacizumab (15 mg/kg) given on day 1 every 3 weeks. The total dose of bevacizumab should only be readjusted according to the current body weight if the body weight changes by more than 10%, as compared to baseline (excluding changes due to development or drainage of ascites, pleural effusions, or fluid retention). Bevacizumab may be omitted from cycle one as specified in Section 5.17. Drugs must be administered in the following order: Bevacizumab 15 mg/kg IV o First cycle infused over 90 minutes o If no problems, then second cycle infused over 60 minutes o If no problems, then third and subsequent cycles infused over 30 minutes Hypersensitivity prophylaxis Antiemetics Paclitaxel infusion 175 mg/m2 over 3 hours (maximum BSA 2.0m2) Carboplatin AUC 6 over 30-60 minutes Post-chemotherapy antiemetics Then: 12 cycles of bevacizumab 15 mg/kg IV over 30 minutes (or fastest rate given previously) given on day 1 every 3 weeks 5.24 Arm IV--Bevacizumab When Given With Oxaliplatin + Capecitabine: Six cycles of chemotherapy (See Section 5.22) plus bevacizumab (15 mg/kg) given on day 1 every three weeks. The total given dose of bevacizumab should only be readjusted according to the current body weight if the body weight changes by more than 10%, as compared to baseline (excluding changes due to development or drainage of ascites, pleural effusions, or fluid retention). Bevacizumab may be omitted from cycle one as specified in Section 5.17. Drugs should be administered in the following order: Bevacizumab 15 mg/kg IV o First cycle infused over 90 minutes -27- GOG-0241 o If no problems, then second cycle infused over 60 minutes o If no problems, then third and subsequent cycles infused over 30 minutes Anitemetics Oxaliplatin 130 mg/m2 over 2-6 hours (maximum BSA = 2.0m2) Capecitabine 850 mg/m2 orally twice daily Post-chemotherapy antiemetics Then: 12 cycles of bevacizumab 15 mg/kg IV over 30 minutes (or fastest rate given previously) given on day 1 every 3 weeks 5.3 Supportive Care Guidelines for Bevacizumab: Prior to each treatment, the patient should be carefully assessed with special attention to blood pressure, proteinuria, bleeding and cardiovascular events. Decisions for retreatment/interruption should follow the dose modification guidelines in Section 6.0. The initial dose will be delivered over a minimum of 90 minutes. If the first infusion is tolerated without infusion-associated adverse events, the second infusion may be delivered over a minimum of 60 minutes. If the 60-minute infusion is well tolerated, all subsequent infusions may be delivered over a minimum of 30 minutes. If an infusion reaction occurs, subsequent doses of bevacizumab should be administered over the shortest period that was well tolerated. 5.331 If a subject experiences a bevacizumab infusion-associated adverse event, such as Grade 1 anaphylactoid reaction, she may be premedicated for the next bevacizumab infusion (see details below in bullet points). Patients who experience a Grade 2 or greater bevacizumab infusionassociated adverse event will not receive any additional bevacizumab. Anaphylaxis precautions should be observed during bevacizumab administration (See Appendix IV). In the event of a prior bevacizumab hypersensitivity reaction, the following prophylactic regimen is recommended upon re-exposure to bevacizumab: H1 blocker (diphenhydramine 25-50 mg IV or orally one hour prior to infusion; or an equivalent dose of an alternate H1 blocker) H2 blocker (ranitidine 50 mg IV or 150 mg orally one hour prior to infusion; or an equivalent dose of an alternate H2 blocker) Dexamethasone (Suggested dosing for dexamethasone, if needed prior to bevacizumab administration, is 10 mg administered orally or 20 mg IV prior to bevacizumab infusion) Acetaminophen for symptom control or premedication as needed. -28- GOG-0241 5.332 Hypertension: Hypertension is a known and potentially serious adverse event associated with bevacizumab treatment. Patients receiving bevacizumab should have their blood pressure monitored weekly during the first cycle of therapy, and subsequently, prior to each infusion of bevacizumab. Home monitoring is permitted. Patients who elect to monitor blood pressure at home will be provided with a Blood Pressure Check Calendar (Appendix V). Patients who monitor their blood pressure at home should be instructed to call their doctor if the systolic blood pressure is >140 mmHg or the diastolic blood pressure is > 90 mmHg. Hypertensive medication should be initiated or increased per routine practice. Bevacizumab treatment modifications due to hypertension should follow the instructions in section 6.0. 5.333 Therapeutic anticoagulation: Patients on therapeutic anticoagulation should have PT/INR or PTT (whichever is appropriate) monitored closely during bevacizumab therapy. Bevacizumab should be held if the coagulation parameters are higher than the intended therapeutic range. 5.334 Wound complications and surgery: The appropriate interval from discontinuation of bevacizumab to subsequent elective surgery required to reduce the risk of impaired wound healing has not been determined. Decision on such an interval should take into consideration the half-life of bevacizumab. It is generally recommended that bevacizumab should be discontinued at least 4 weeks prior to major elective surgery. In addition, bevacizumab should not be restarted until at least 4 weeks after major surgery provided that the wound has adequately healed. 5.335 Re-imaging of measurable disease sites by CT scan of the chest/abdomen/pelvis will be performed prior to every other cycle. Patients who have complete response (CR), partial response (PR), or stable disease will continue for additional cycles. Patients who have progression of disease will be removed from study treatment. If the patient’s response is CR, PR, or stable disease, and if side effects are not severe, a patient may remain on a study agent indefinitely at the investigator's discretion. 5.336 Chemotherapy Guidelines If a patient receiving chemotherapy (with or without bevacizumab) experiences a > 10% weight change, the chemotherapy dose to be administered with a subsequent cycle must be recalculated. For 21 day cycles, a patient will be permitted to have a new cycle of chemotherapy delayed up to 7 days (without this being considered to be a protocol violation) for major life events (e.g., serious illness in a family member, vacation which is unable to be re-scheduled). Documentation to justify this decision should be provided in the physician’s clinic note. It will be acceptable for individual chemotherapy doses to be delivered within a “24-hour window before and after the protocol-defined date” for -29- GOG-0241 21 day cycles. If the treatment due date is a Friday, and the patient cannot be treated on that Friday, then the window for treatment would include the Thursday (1 day earlier than due) through the Monday (day 3 past due). Chemotherapy doses can be “rounded” to +/- 5% of the calculated dose without being considered a protocol violation. Please note: If bevacizumab is interrupted for ANY reason for > 8 weeks, the patient should discontinue bevacizumab therapy on protocol. Patients will continue to receive carboplatin/paclitaxel and oxaliplatin/capecitabine on study. 5.337 Patients who have an ongoing study agent-related serious adverse event upon study completion or at discontinuation from the study treatment will be contacted by the investigator or his/her designee periodically until the event is resolved or determined to be irreversible. 5.4 Dexamethasone and G-CSF (Neupogen) Compliance The use of a Patient Pill Calendar (see Appendix V) during study therapy will be utilized by the patient and the treating clinic to help promote and monitor compliance with dexamethasone. Patients who are treated with G-CSF (Neupogen) or Neulasta at home will also record G-CSF use on this calendar. 5.5 Blood Pressure Check Compliance The use of a Blood Pressure Check Calendar (see Appendix VI) during study therapy will be utilized by the patient whenever blood pressure is taken at home to help promote and monitor compliance with this study parameter. Patients may elect to check their blood pressure at home if they have access to a blood pressure monitor (home blood pressure monitors will not be provided by the study). Alternatively, blood pressure checks may be done in other outside settings, such as a pharmacy or outside physician’s office, with results recorded on the Blood Pressure Check Calendar. 5.6 Criteria for removal from treatment 5.61 Inability to tolerate the lowest doses because of toxicity. 5.62 The patient may withdraw from the study at any time for any reason. 5.63 Patients with evidence of progressive disease or other patients with significant side effects or deterioration of performance status may be removed from study at the investigator's discretion. -306.0 GOG-0241 TREATMENT MODIFICATIONS In order to maintain dose-intensity and cumulative dose-delivery on this study, reasonable efforts will be made to minimize dose reduction and treatment delays as specified. Any patient whose treatment is delayed must be evaluated on a weekly basis until adequate hematologic and non-hematologic parameters have been met. No dose escalation is planned for this study. 6.1 Arm I: Paclitaxel + Carboplatin: 6.11 General Guidelines for Hematologic Toxicity 6.111 Initial treatment modifications will consist of cycle delay and/or dose reduction as directed. 6.112 Treatment decisions will be based on the absolute neutrophil count (ANC) rather than the total white cell count (WBC). 6.113 Subsequent cycles of therapy will not begin until the ANC > 1500 cells/mm3 (CTC Grade 1) and the platelet count is > 75,000/mm3 (CTC Grade 1). Therapy will be delayed for a maximum of three weeks until these values are achieved. Patients who fail to recover adequate counts within a threeweek delay will no longer receive protocol-directed chemotherapy. Patients who received G-CSF prior to the current cycle may begin with ANC > 1000 cells/mm3, if clinically appropriate, to allow for transient reductions in ANC after discontinuation of G-CSF. Patients who are delayed more than 7 days may begin with ANC > 1000 cells/mm3, if clinically appropriate, as they will receive G-CSF with subsequent therapy. 6.114 The use of hematopoietic cytokines and protective reagents are restricted as noted: 6.1141 In general, patients will NOT receive prophylactic filgrastim (GCSF), PEG-filgrastim (Neulasta), or sargramostim (GM-CSF) unless they experience treatment delays or recurrent neutropenic complications after treatment modifications as specified. In particular, hematopoietic growth factors should NOT be used to avoid initial chemotherapy dose modifications as stipulated in the protocol. However, patients may receive growth factors for management of neutropenic complications in accordance with clinical treatment guidelines. If required, it is recommended that growth factors be initiated the day after the last dose of chemotherapy. NeupogenTM should continue for a minimum of 10 days or until the ANC is sustained above > 1000/mm3. Growth factors should be discontinued if the ANC exceeds 10,000/mm3 and should not be used within 72 hours of a subsequent dose of chemotherapy. -31- GOG-0241 6.1142 Patients should NOT receive prophylactic thrombopoietic agents unless they experience recurrent grade 4 thrombocytopenia after treatment modifications as specified below. 6.1143 Patients may receive erythropoietin (EPO), iron supplements, and/or transfusions as clinically indicated for management of anemia, but should NOT receive EPO when the Hb is > 10.0 g/dl. 6.1144Patients may NOT receive amifostine or other protective reagents unless indicated in the study design. 6.12 Modifications for Hematologic Toxicity (Nadirs) 6.121 Initial occurrences of dose-limiting neutropenia (defined in 6.122) and dose-limiting thrombocytopenia (defined in 6.223) will be performed according to Table A, using the regimen modifications in Table B. 6.122 Dose-limiting neutropenia (DLT-ANC) is defined by the occurrence of febrile neutropenia or grade 4 neutropenia. Febrile neutropenia is defined within the CTC as fever of unknown origin without clinically or microbiologically documented infection with ANC less than 1,000 cells/mm3 and fever greater than or equal to 38.5C. This is to be distinguished from catheter-related infections and other documented infections that occur within the setting of grade 3-4 neutropenia. 6.123 Dose-limiting thrombocytopenia (DLT-PLT) is defined by any occurrence of grade 4 thrombocytopenia (< 10,000/mm3) or bleeding associated with grade 3 thrombocytopenia (10,000 to < 50,000/mm3). There will no modifications for uncomplicated grade 3 thrombocytopenia. Table A: DLT DLT ANC PLT Yes No Yes Yes No Yes Table B: Modification Instructions for Dose-Limiting Hematoloigic Toxicity (In conjunction with Table B) First Occurrence Second Occurrence Third Occurrence Reduce the regimen drug doses by one level using Table B. Reduce the regimen drug doses by one level using Table B. For carboplatin AUC 6, decrease one AUC unit and maintain other drug doses. Add G-CSF and maintain all drug doses. Add G-CSF and decrease carboplatin one AUC unit. For carboplatin AUC 5, decrease one AUC unit and maintain other drug doses. Off Study Treatment Off Study Treatment Off Study Treatment Regimen Modifications for Hematologic Toxicity (In conjunction with Table A -32Drug Carboplatin Paclitaxel Regimen -2 Level AUC 4 175 mg/m2 6.13 Regimen -1 Level AUC 5 175 mg/m2 GOG-0241 Regimen Starting Dose Level AUC 6 175 mg/m2 Modifications for delayed hematologic recovery 6.131 Delay on the basis of neutropenia (Delay-ANC) is defined if the ANC is less than 1500 cells/mm3 (CTC grade 2 or worse) within 24 hours prior to scheduled therapy, or less than 1000 cells/mm3, if the patient received G-CSF during the previous cycle. 6.132 Delay on the basis of thrombocytopenia (Delay-PLT) is defined if the platelet count is less than 75,000/mm3 (CTC grade 2 or worse) within 24 hours prior to scheduled therapy. 6.133 Modifications noted below are only required for management of delays in the absence of dose reductions stipulated by nadir DLT-ANC and/or DLT-PLT (as noted above). In other words, if the patient experiences DLT-ANC and Delay-PLT, make the modifications as indicated for the nadir counts without additional modifications based on delayed recovery. Table C: Category Delay-ANC Delay-PLT Modifications for Delayed Hematologic Recovery Delay (days) 1-7 8-21 >21 1-7 8-21 >21 6.14 Modification No change Add G-CSF with next cycle Off study treatment No change Decrease carboplatin one AUC unit, but not lower than AUC 4 Off study treatment Modifications for Non-Hematologic Toxicity Table D: Drug Carboplatin Paclitaxel -2 Level AUC 5 110 mg/m2 Allowable Dose Levels for Individual Drugs -1 Level AUC 6 135 mg/m2 0 Level AUC 6 175 mg/m2 6.141 Grade 2 peripheral neuropathy requires reduction of one dose level in paclitaxel and delay in subsequent therapy for a maximum of 3 weeks until recovered to grade 1. For grade > 3 neurotoxicity, paclitaxel should be permanently discontinued and subjects treated with single-agent carboplatin with no dose reduction. -33- GOG-0241 6.142 Renal toxicity associated with reduction in GFR is not expected as a direct complication of chemotherapy in this untreated patient population using the prescribed dose and schedule of this regimen. As such, there are no specific dose modifications for renal toxicity. However, the target AUC dose of carboplatin must be recalculated each cycle in any patient who develops renal toxicity, defined by serum creatinine greater than 1.5 x institutional upper limit normal (ULN), CTCAE Grade > 2. 6.143 Hepatic toxicity is not expected as a direct complication of this regimen in this untreated patient population using the prescribed dose and schedule. However, the development of Grade 3 (or greater) elevations in SGOT (AST), SGPT, (ALT), alkaline phosphatase or bilirubin requires reduction of one dose level in paclitaxel and delay in subsequent therapy for a maximum of three weeks until recovered to Grade 1. 6.144 There will be no dose modifications for alopecia, constipation, or nausea. It is recommended that routine medical measures be employed to manage nausea, and constipation. Note: Once the dose has been reduced, it should not be increased at a later time. 6.2 Arm II: Oxaliplatin + Capecitabine Toxicity Myelosuppression Grade 0 or 1 Grade 2 Grade 3 Capecitabine Oxaliplatin Maintain dose Decrease 1 D.L Delay dose, then decrease 1 D.L. when resolved to < gr 1 Maintain dose Maintain dose Maintain dose for neutropenia; for thrombocytopenia delay dose then decrease 1 D.L. Grade 4 Delay dose, then decrease 1 D.L. when resolved to < gr 1 Delay dose then decrease 1 D.L. when resolved to ANC>1500 Febrile Neutropenia Delay dose, then decrease 1 D.L. when resolved Delay dose, then decrease 1 D.L. when resolved Diarrhea Grade 0 Grade 1 Grade 2 or 3 Maintain dose Maintain dose Delay dose then resume Delay then maintain dose at same dose when resolved Delay dose, then decrease 1 D.L. when resolved Delay then maintain dose -34Grade 4 Mucositis Grade 0 or 1 Grade 2 Grade 3 Grade 4 GOG-0241 Delay dose, then Delay then maintain dose decrease 2 D.L. when resolved or discontinue at investigator discretion Maintain dose Decrease 1 D.L Delay dose, then decrease 1 D.L. when resolved Maintain dose Maintain dose Delay then maintain dose Delay dose, then Delay then maintain dose decrease 2 D.L. when resolved or discontinue at investigator discretion 1 Dose level (D.L.) = 75% of initial dose; 2 Dose levels (D.L.) = 50% of initial dose Toxicity Vomiting Grade 0 or 1 Grade 2 Capecitabine Oxaliplatin Maintain dose Delay dose, then decrease 1 D.L. when resolved Maintain dose Delay then Maintain dose Grade 3 Delay dose, then decrease 1 D.L. when resolved Delay dose, then Maintain dose; consider modification of antiemetic schedule Grade 4 Delay dose, then Delay dose, then decrease 1 D.L. decrease 2 D.L. when when resolved to < gr 1 resolved or discontinue at investigator discretion Neuropathy/Parasthesia Dysethesias with cold Parasthesias Parasthesias with pain < 7 days Maintain dose Maintain dose Maintain dose Maintain dose Maintain dose Maintain dose Parasthesias with pain > 7 days (normal exam) Maintain dose Reduce 1 D.L. Parasthesias with pain > 7 days (abnormal exam) Maintain dose Omit oxaliplatin one cycle then restart with a reduction of 1 D.L. -35Parasthesias with pain: Persistent Maintain dose GOG-0241 Stop until improvement. If improvement, restart with a reduction of 1 D.L. Parasthesias with functional Maintain dose impairment <7 days Maintain dose Parasthesias with functional Maintain dose impairment >7 days (normal exam) Normal exam and not interfering with ADL's: Reduce 1 D.L. Parasthesias with functional Maintain dose impairment >7 days (abnormal exam) Omit oxaliplatin one cycle then restart with a reduction of 1 D.L. Parasthesias with functional Discontinue protocol impairment - Persistent Discontinue protocol 1 Dose level (D.L.) = 75% of initial dose; 2 Dose levels (D.L.) = 50% of initial dose Toxicity Hand-Foot Syndrome Grade 0 or 1 Grade 2 (1st appearance) Grade 2 (2nd appearance) Grade 3 (1st appearance) Grade 3 (2nd appearance) Grade 4 Capecitabine Maintain Dose Maintain Dose Decrease 1 D.L. Decrease 1 D.L. Decrease 2 D.L. Discontinue protocol Thromboembolic Events Grade 0,1, or 2 Maintain dose Grade 3 or 4 Delay chemotherapy, start anticoagulation, resume at investigator discretion Oxaliplatin Maintain Dose Maintain Dose Maintain Dose Maintain Dose Maintain Dose Discontinue protocol Maintain dose Delay chemotherapy, start anticoagulation, resume at investigator discretion All other nonhematological toxicities, except alopecia Grade 0 or 1 Grade 2 Maintain dose Delay dose, then decrease 1 D.L. when resolved Maintain dose Maintain dose -36- GOG-0241 Grade 3 Delay dose, then decrease 1 D.L. when resolved Delay dose, then decrease 1 D.L. when resolved Grade 4 Delay dose, then Delay dose, then decrease 1 D.L. decrease 2 D.L. when when resolved resolved or discontinue at investigator discretion 1 Dose level (D.L.) = 75% of initial dose; 2 Dose levels (D.L.) = 50% of initial dose Note: Dose delays can be for up to a maximum of three weeks. Laryngopharyngeal dysesthesia If laryngopharyngeal dysesthesia occurs, the next dose of oxaliplatin should be administered as a six-hour infusion. Cold exposure should be strictly avoided. 6.3 Bevacizumab Treatment Modification Criteria and Guidelines for Management Event Allergic reactions or Acute infusional reactions/cytokine release syndrome CTCAE.v3.0 Action to be Taken Grade Grade 1-3 If infusion-related or allergic reactions occur with bevacizumab, premeds should be given with the next dose, and infusion time may not be reduced for the subsequent infusion. Follow the guidelines in Section 5.2 for bevacizumab administration. For patients with Grade 3 reactions, bevacizumab infusion should be stopped and not restarted on the same day. At the physicians’ discretion, bevacizumab may be permanently discontinued or re-instituted with premeds and at a rate of 90+15 min. If bevacizumab is re-instituted, the patient should be closely monitored for a duration comparable to or longer than the duration of the previous reactions. Discontinue bevacizumab Discontinue bevacizumab. Grade 4 Arterial Thrombosis Grade 2 ( if - Cardiac ischemia/ new or infarction worsened since - CNS ischemia bevacizumab (TIA, CVA) therapy) - any peripheral or Grade 3-4 Discontinue bevacizumab visceral arterial ischemia/throm- -37- GOG-0241 bosis Venous Thrombosis Grade 3 OR asymptomatic Grade 4 Venous Thrombosis (continued) Hold bevacizumab treatment. If the planned duration of full-dose anticoagulation is <2weeks, bevacizumab should be held until the full-dose anticoagulation period is over. If the planned duration of full-dose anticoagulation is >2 weeks, bevacizumab may be resumed during the period of full-dose anticoagulation IF all of the criteria below are met: - The subject must have an in-range INR (usually 2-3) on a stable dose of warfarin or on a stable dose of heparin prior to restarting bevacizumab. - The subject must not have pathological conditions that carry high risk of bleeding (e.g. tumor involving major vessels or other conditions). - The subject must not have had significant hemorrhagic events while on study. If thromboemboli worsen/recur upon resumption of study therapy, discontinue bevacizumab Discontinue bevacizumab Hypertension Proteinuria Grade 4 (symptomatic) [Treat with anti-hypertensive medication as needed. The goal of BP control should be consistent with general medical practice] Controlled BP at Continue bevacizumab the discretion of the treating physician Persistent or Hold bevacizumab. If treatment is delayed symptomatic for > 8 weeks due to uncontrolled HTN hypertension, discontinue bevacizumab. Grade 4 Discontinue bevacizumab. [Proteinuria should be monitored by urine analysis for urine protein creatinine (UPC) ratio prior to every other dose of bevacizumab] UPC ratio Continue bevacizumab. < 3.5 -38UPC ratio > 3.5 Grade 4 or nephrotic syndrome Wound dehiscence requiring medical or surgical intervention GI perforation, GI leak or fistula Other clinically significant AEs attributable to bevacizumab (except controlled nausea/vomiting). Grade 3 Hold bevacizumab until UPC recovers to < 3.5. If therapy is held for > 2 months (8 weeks) due to proteinuria, discontinue bevacizumab. Discontinue bevacizumab. Discontinue bevacizumab Discontinue bevacizumab Grade 4 GOG-0241 Hold bevacizumab until symptoms resolve to < grade 1 If treatment delay is >3-4 weeks due to toxicity, discontinue bevacizumab. Discontinue bevacizumab Upon consultation with the study chair, resumption of bevacizumab may be considered if a patient is benefiting from therapy, and the G4 toxicity is transient, has recovered to < grade 1 and unlikely to recur with retreatment. *Current CTCAE definitions used by CTEP: Grade 1: asymptomatic, transient (< 24 hours) increase by > 20 mmHg (diastolic) or to >150/100 if previously WNL; intervention not indicated Grade 2: recurrent or persistent (> 24 hours) or symptomatic increase by > 20 mmHg (diastolic) or to > 150/100 if previously WNL; monotherapy may be indicated Grade 3: requiring more than one drug or more intensive therapy than previously Grade 4: life threatening (e.g. hypertensive crisis) Patients who interrupt or discontinue bevacizumab for bevacizumab-related toxicity should continue on-time treatment with gemcitabine and docetaxel. Imaging (CT scans) should continue to be performed on time in order to determine if there is evidence of progression of disease. 6.4 Dose escalations There will be no dose escalations or re-escalations on this study. -397.0 GOG-0241 STUDY PARAMETERS 7.1 Observations and Tests The following observations and tests are to be performed and recorded on the appropriate form(s) -40- Prior to Study Therapy Prior to each cycle for cycles 1-6 History & Physical 1 Blood pressure 1 Coagulation parameters (INR, APTTN) 2 Toxicity Assessment 2 Serum pregnancy test 3 CBC with Diff, Platelets Between cycles 3 and 4, and 1 month post cycle 6 GOG-0241 Post Cycle 6 (weeks 18-54). Post Treatment No Bevacizumab† 12 cycles of Bevacizumab (Cycles 7-18) ‡ q 3 months in Year 2 then q 6 months in Years 3-5, then annually X X X X X X X X X X11 X X 2 X* X X 5 Lytes, BUN, Creat, Ca, Mg, PO4, Bili, SGOT, Alk Phos, Total protein, albumin 2 X X X 5 EKG 1 CT Scan of chest X1 X Radiographic Tumor Measurement (MRI or CT abdomen/pelvis) X1 X 6, 8 6, 8 7, 8 CA-125, Carcinoembryonic Antigen (CEA), CA19-9 X2 X X X X Colonoscopic colon cancer screening 4 Urinalysis 2 X X X FACT-TOI/Ntx-4 and EQ-5D™ X 9 9 PARAMETER X 10 Telephone Call 11 If grade 4 neutropenia is documented (ANC<500/mm3), obtain weekly until resolved to grade 3. Only if the initial CT scan of the chest is abnormal † Assessments via clinical visits every 3 months and telephone calls every 3 weeks between clinic visits. ‡ Assessments every 3 weeks unless otherwise specified. 1. Must be obtained within 28 days prior to initiating protocol therapy. Patients undergoing surgery need a scan of the abdomen/pelvis pre- and post-surgery. The same scanning technique must be used throughout the whole trial. 2. Must be obtained within 14 days prior to initiating protocol therapy. Urine dipstick for proteinuria <2+. If urine dipstick is >2+ on two occasions more than one week apart, then a 24-hour urine must demonstrate <1g of protein in 24 hours. For patients on full anticoagulation, refer to Section 6.3 for appropriate management. 3. Must be obtained within 72 hours prior to initiating protocol therapy. 4. Must have within 1 year of enrolling on protocol. Patients with bowel resection at time of debulking and without prior colonoscopy may have colonoscopy 4 weeks after surgery and prior to enrolling on protocol. 5. As clinically indicated. 6. CT or MRI scan of abdomen and pelvis (MRI pelvis optional) every 3 months. Imaging can be obtained with asymptomatic CA125 rise in accordance to local practice. -41- GOG-0241 7. CT or MRI scan of abdomen and pelvis (MRI pelvis optional) every 6 months during year 2 and every 12 months during years 3-5. Imaging can be obtained with asymptomatic CA125 rise in accordance to local practice. 8. Patients with measurable disease and a newly documented response (CR or PR) at the conclusion of therapy should undergo repeat imaging in at least 4 weeks (but within 3 months) to confirm the persistence of response in accordance with RECIST version 1.1. 9. QOL questionnaires to be completed: 6 months after end of chemotherapy after completion of maintenance bevacizumab therapy (or 36 weeks after completion of chemotherapy in the non-bevacizumab arms). 10. QOL questionnaires to be completed: 6 months after completion of bevacizumab (or 14 months after completion of chemotherapy) every 12 months in years 2-5. 11. Telephone Assessments: This applies to patients who are not receiving bevacizumab During weeks 18-54, patients who are not receiving bevacizumab will be seen every 3 months. Every 3 weeks, between each 3-month visit, the research nurse must make a telephone call to the patient (Please note: this duty must be signed off on the delegation of responsibilities) The following questions must be asked: How have you been feeling in general? Have you noticed any new symptoms, discomfort or pain? Has there been any change to your existing symptoms? Have there been any changes to your medication? Answers must be acted on accordingly, as they would be if the patient were in clinic. Blank CRFs will be provided for completion, to record the content of the telephone call. These must be completed by the research nurse, and a copy returned to the GOG Statistical Center. 7.2 Pathology Requirements The histopathologic diagnosis of primary advanced stage mucinous adenocarcinoma is made based on pathology review at each institutional site. To verify that patients on this protocol actually have a primary mucinous ovarian carcinoma, one block should be taken for every centimeter diameter of the tumor, taking the maximum dimension as the diameter. Sampling should concentrate particularly on solid areas and potential areas of capsular involvement by tumor. Where the ovarian hilum can still be identified, it should be sampled in particular for the detection of vessel invasion. As an entry requirement, immunohistochemistry must be performed upon a section of the primary tumor with antibodies directed against CK7, CK20, and CDX2. (Whenever possible, immunostaining with monoclonal CEA, CA 19-9, CA 125, estrogen receptor (ER), and progesterone receptor (PR), is recommended, but it is not required for patient entry). In addition to the surgical pathology report, all H&E stained slides of the ovarian tumor,all immunostained slides and one slide to show the most advanced stage of disease must be submitted for central review by the two study pathologists. Slides from recurrence and/or persistent disease are required only for Stage I tumors, when recurrence/persistent disease is the basis for eligibility. In addition to the immunohistochemical results, it is recommended that the institutional pathologists consider the WHO and other publications that have identified criteria that help to distinguish primary mucinous carcinomas of the ovary from those that are -42- GOG-0241 metastatic to the ovary from other sites. Features suggestive of metastasis follow, and the datasheet with this information must be completed and sent by the institutional pathologist: 1) bilaterality 2) a nodular pattern of ovarian involvement 3) an infiltrative pattern of stromal invasion 4) microscopic surface deposits of tumour 5) modest sized ovarian tumours 6) extensive lymphovascular invasion, especially in the hilum and perihilar tissues 7) single cell infiltration 8) signet ring differentiation 9) mucin pooling with individual cells floating in the mucinous material 10) marked pattern variation in tumour from one nodule to another 11) immunohistochemical phenotype showing stronger cytokeratin 20 than 7 positivity 12) particularly for metastatic bowel tumour, the presence of dirty necrosis, segmental necrosis and surviving ribbons of glandular cells in a garland-like pattern. 13) a florid stromal luteinised cellular response Any of these features may also be seen in primary ovarian mucinous carcinomas. However, the chance of a tumor being a metastasis is increased when these features are present in combination. When submitting pathology material to the GOG SDC, individual slides must be labeled with GOG Patient ID, patient initials and the surgical / pathology accession number (e.g., S08-2355) and block identifier (e.g., A6). Don’t not label with disease site or collection date. Pack the labeled slides into plastic slide cassette(s). Tape plastic slide cassettes shut and wrap in bubble wrap or another type of padded material prior to shipping. Please include the GOG Patient ID, patient initials, and protocol number on all pages of the pathology report and black out the patient’s name. Ship pathology slides, three copies of both the Pathology Form F and the official pathology report directly to the Pathology Materials Coordinator at the GOG Statistical and Data Center, Roswell Park Cancer Institute, Research Studies Center, Carlton and Elm Streets, Buffalo, New York, 14263; phone (716) 845-5702. The GOG Upload Application is an alternative method for submitting stained slides, pathology reports and Form F. Central pathology review by two GOG pathologists will be required but not prior to enrollment on this protocol, and is not the basis for determination of eligibility for entry on this protocol. This review will include study of H & E sections and immunostaining, with careful adherence to the distinction between primary versus metastatic ovarian involvement based on WHO criteria. 7.3 Specimen Requirements and Laboratory Testing Specimen (Specimen Code) Collection Time Point Shipping -43Formalin-fixed, paraffin-embedded primary tumor tissue (a block or 15 charged slides) (FP01) Formalin-fixed, paraffin-embedded primary tumor tissue (a 50-micron curl) (FP02) 7.31 Archival GOG-0241 Ship at room temperature to the GOG Tissue Bank using your own container Archival VEGF/EGF Ligand, Receptor, and Activated Receptor Immunohistochemistry 7.311 A block or fifteen serial sections from archived formalin-fixed, paraffinembedded primary tumor tissue should be obtained from your pathology department. Serial sections will be placed on charged slides suitable for immunohistochemistry (IHC). Slides will be used in IHC assays to determine protein levels of VEGF, EGF, VEGF receptor, EGF receptor, and phosphorylated EGF-R and VEGF-R. 7.32 KRAS DNA Analysis 7.321 A 50 micron curl from fixed and embedded primary tumor will also be required. Isolation of DNA and direct sequence analysis of KRAS will be performed as previously described in detail.31 7.4 Quality of Life Assessments Quality of life (QOL) assessments will include the FACT-TOI (26 questions), FACT-GOG/NTX-4 (4 questions), and EQ-5D™ (6 questions) The time to complete these surveys should be 5 minutes or less based on prior quality of life studies. They will be administered to the patient as a self-report assessment. Patients will be instructed to read the brief instructions at the top of the form, and after the patients’ understanding of the instructions has been confirmed, the patient should be instructed to complete every item in order, without skipping any items. If a patients’ family member remains with the patient while she is completing the surveys, the data managers should encourage patients to answer the questions on their own. If patients are unable to come to the clinic, the questionnaire may be mailed to them with a request to complete it at home and to return it via mail to the appropriate individual using a self-addressed stamped envelope provided by the study team. This request should be followed by a telephone call by the data manager to make sure the patient has received the materials and to ensure completion of the material. -448.0 GOG-0241 EVALUATION CRITERIA 8.1 Parameters of Response – RECIST (version 1.1) Criteria56 8.11 Measurable disease (“Target”) is defined as at least one lesion that can be accurately measured in at least one dimension (longest diameter to be recorded). Each lesion must be 10 mm when measured by CT (CT scan slice thickness no greater than 5 mm*); > 10 mm caliper measurement by clinical exam (lesions which cannot be accurately measured with calipers should be recorded as non-measurable); and > 20 mm by chest x-ray. *If CT scan with slice thickness > 5 mm is used, the minimum lesion size must have a longest dimension twice the actual slice thickness. Malignant lymph nodes: To be considered pathologically enlarged and measurable, a lymph node must be > 15 mm in short axis when assessed by CT scan (CT scan slice thickness recommended to be no greater than 5 mm). At baseline and in follow-up, only the short axis will be measured and followed. Clinical lesions will only be considered measureable when they are superficial and > 10 mm diameter as assessed using calipers (e.g. skin nodules). When lesions can be evaluated by both clinical exam and imaging, imaging evaluation should be undertaken since it is more objective and my also be reviewed at the end of the study. CT is the best currently available and reproducible method to measure lesions selected for response assessment. MRI may be substituted for contrast enhanced CT for some sites (e.g. for abdomen and/or pelvis), but NOT lung. The minimum size for measurability is the same as for CT (10 mm) as long as the scans are performed with slice thickness of 5 mm and no gap. In the event the MRI is performed with thicker slices, the size of a measurable lesion at baseline should be two times the slice thickness. Tumors within a previously irradiated field will be designated as “nontarget” lesions unless progression is documented or a biopsy is obtained to confirm persistence at least 90 days following completion of radiation therapy. Bone lesions: Bone scan, PET scan or plain films are NOT considered adequate imaging techniques to measure bone lesions. Lytic bone lesions or mixed lytic-blastic lesions, with identifiable soft tissue components, that can be evaluated by cross sectional imaging techniques such as CT or MRI can be considered as measurable if the -45- GOG-0241 soft tissue component meets the definition of measurability described above. Blastic bone lesions are non-measurable. Cystic lesions: Lesions that meet the criteria for radiographically defined simple cysts should not be considered as malignant lesions (neither measurable nor non-measurable). Cystic lesions thought to represent cystic metastases can be considered measurable lesions, if they meet the definition of measurability described above. However, if non-cystic lesions are present in the same patient, these are preferred for selection as target lesions. 8.12 Non-measurable disease (“Non-Target”) is defined as all other lesions, including small lesions (<10 mm or pathological lymph nodes with > 10 to < 15 mm short axis) as well as truly non-measurable lesions. Lesions considered truly non-measurable include: Leptomeningeal disease Ascites Pleural or pericardial effusion Inflammatory breast disease Lymphangitic involvement of skin or lung Abdominal masses/abdominal organomegaly indentified by physical exam that is not measureable by reproducible imaging techniques 8.13 Baseline documentation of “Target” and “Non-Target” lesions All measurable lesions up to a maximum of 5 lesions total (and a maximum of two lesions per organ) representative of all involved organs should be identified as target lesions and recorded and measured at baseline. Target lesions should be selected on the basis of their size (lesions with the longest diameter), be representative of all involved organs, but in addition should be those that lend themselves to reproducible repeated measurements. A sum of the diameters (longest for non-nodal lesions, short axis for nodal lesions) for all target lesions will be calculated and reported as the baseline sum diameters. The baseline sum diameters will be used as reference to further characterize the objective tumor response of the measurable dimension of the disease. All other lesions (or sites of disease) should be identified as non-target lesions and should also be recorded at baseline. Measurements are not required and these lesions should be followed as “present” or “absent”. It is possible to record multiple non-target lesions involving the same organ -46- GOG-0241 as a single item on the D2M form (e.g. ‘multiple enlarged pelvic lymph nodes’ or ‘multiple liver metastates”). All baseline evaluations of disease status should be performed as close as possible to the start of treatment and never more than 4 weeks before the beginning of the treatment. The same method of assessment and the same technique should be used to characterize each identified and reported lesion at baseline and during follow-up. Imaging based evaluation should ALWAYS be done rather than clinical examination unless the lesion(s) being followed cannot be imaged but are assessable by clinical exam. 8.14 Response Criteria 8.141 Complete Response (CR): Disappearance of all target and nontarget lesions. Any pathological lymph nodes (whether target or non-target) must have reduction in short axis to <10 mm. Normalization of CA125, if elevated at baseline, is required for ovarian/primary peritoneal/fallopian tube cancer studies. 8.142 Partial Response (PR): At least a 30% decrease in the sum of diameters of target lesions, taking into reference the baseline sum diameters. 8.143 Progressive Disease (PD): At least a 20% increase in the sum of diameters of target lesions, taking as reference the smallest sum on study (this includes the baseline sum if that is the smallest on study). In addition to the relative increase of 20%, the sum must also demonstrate an absolute increase of at least 5 mm. (Note: the appearance of one or more NEW lesions is also considered progression. Guidance on when a lesion is to be considered new is provided in the above cited reference). Unequivocal progression of existing non-target lesions is also considered progression (a detailed description and examples of unequivocal progression of existing non-target lesions is provided in the above cited reference). For equivocal findings of progression (e.g. very small and uncertain new lesions; cystic changes or necrosis in existing lesions), treatment may continue until the next scheduled assessment. If at the next scheduled assessment, progression is confirmed, the date of progression should be the earlier date when progression was suspected. -47- GOG-0241 8.144 Stable Disease (SD): Neither sufficient shrinkage to qualify for PR nor sufficient increase to qualify for PD, taking as reference the smallest sum diameters while on study. 8.145 Not evaluable (NE) is when no imaging/measurement is done at all at a particular time point. The patient is not evaluable (NE) at that time point. 8.146 Early death is defined as having NO repeat tumor assessments following initiation of study therapy resulting from the death of the patient due to disease or treatment. Patients with global deterioration of health status requiring discontinuation of treatment without objective evidence of disease progression at that time will be recorded as ‘symptomatic deterioration’. Every effort should be made to document objective progression even after discontinuation of treatment. Confirmation of response (for both CR and PR): Complete or partial response may only be claimed if the criteria for each are met at a subsequent time point (> 4 weeks later) in studies with a primary endpoint that includes response rate. When response rate is a secondary endpoint (e.g. randomized phase II or III studies with progression-free survival or overall survival as primary endpoint) confirmation is NOT required. Special note on lymph nodes: Lymph nodes identified as target lesions should always have the actual short axis measurement recorded (measured in the same anatomical plane as the baseline examination), even if the nodes regress to below 10 mm on study. This means that when lymph nodes are included as target lesions, the ‘sum’ of lesions may not be zero even if complete response criteria are met, since a normal lymph node is defined as having a short axis of < 10 mm. For PR, SD and PD, the actual short axis measurement of the nodes is to be included in the sum of target lesions. Special note on target lesions that become ‘too small to measure’: While on study, all lesions (nodal and non-nodal) recorded at baseline should have their actual measurements recorded at each subsequent evaluation, even when very small (e.g. 2 mm). However, sometimes lesions or lymph nodes which are recorded as target lesions at baseline become so faint on CT scan that the radiologist may not feel comfortable assigning an exact measure and may report them as being ‘too small to measure’. When this occurs it is important that a value be recorded on the -48- GOG-0241 D2M form. If it is the opinion of the radiologist that the lesion has likely disappeared, the measurement should be recorded as 0 mm. If the lesion is believed to be present and is faintly seen but too small to measure, a default value of 5 mm should be assigned (Note: It is less likely that this rule will be used for lymph nodes since they usually have a definable size when normal and are frequently surrounded by fat such as in the retroperitoneum; however, if a lymph node is believed to be present and is faintly seen but too small to measure, a default value of 5 mm should be assigned in this circumstance as well). 8.15 Evaluation of best overall response is according to Tables 1-3: Table 1 is used for patients with measurable disease at baseline. Table 1: Time point response: patients with target (+/– non-target) disease Target lesions Non-target lesions New lesions Overall response CR CR No CR CR Non-CR/non-PD No PR CR Not evaluated No PR PR Non-PD or not all evaluated No PR SD Non-PD or not all evaluated No SD No NE Not all evaluated Non-PD PD Any Yes or No PD Any PD Yes or No PD Any Any Yes PD Table 2 is used for patients with non-measurable disease. Table 2: Time point response: patients with non-target disease only Non-target lesions New lesions Overall response CR No CR Non-CR/non-PD No Non-CR/non-PD Not all evaluated No NE Unequivocal PD Yes or No PD -49- GOG-0241 Non-target lesions New lesions Overall response Any Yes PD Table 3 is used for phase II trials studies with a primary endpoint that includes response rate. Table 3: Best overall response when confirmation of CR and PR required Overall response Overall response First time point Subsequent time point BEST overall response CR CR CR CR PR SD, PD or PR* CR SD SD CR PD SD CR NE SD PR CR PR PR PR PR PR SD SD PR PD SD PR NE SD NE NE NE *If a CR is truly met at first time point, then any disease seen at a subsequent time point, even disease meeting PR criteria relative to baseline, makes the disease PD at that point (since disease must have reappeared after CR). However, sometimes ‘CR’ may be claimed when subsequent scans suggest small lesions were likely still present and in fact the patient had PR or SD, not CR at the first time point. Under these circumstances, the original CR should be changed to PR or SD and the best response is PR or SD. 8.16 Duration of response is defined as the time measurement criteria are first met for CR/PR until the first date that recurrent or progressive disease is objectively documented (taking as reference for progressive disease the smallest measurement recorded on study). 8.17 Duration of stable disease is measured from the start of the treatment (in randomized trials, from the date of randomization) until the criteria for progression are met, taking as reference the smallest sum on study (if the baseline sum is the smallest, this is the reference for calculation of PD). -50- GOG-0241 8.18 Progression-Free Survival is the period from study entry until recurrent or progressive disease is objectively documented (taking as reference for progressive disease the smallest measurement recorded on study), death or date of last contact. 8.19 Survival is the observed length of life from entry into the study to death or the date of last contact. -51- GOG-0241 -529.0 GOG-0241 DURATION OF STUDY 9.1 Patients will continue on study treatment until they complete 6 cycles of chemotherapy or disease progression or adverse effects prohibit further treatment. The patient can refuse the study treatment at any time. 9.2 All patients will be followed (with completion of all required case report forms) until disease progression or study withdrawal. In addition, following disease progression, patients will be monitored for delayed toxicity and survival for a period of 5 years with Q forms submitted to the GOG Statistical and Data Center, unless consent is withdrawn. -53- 10.0 GOG-0241 STUDY MONITORING AND REPORTING PROCEDURES 10.1 ADVERSE EVENT REPORTING FOR A COMMERCIAL AGENT 10.11 Definition of Adverse Events (AE) An adverse event (AE) is any unfavorable and unintended sign (including an abnormal laboratory finding), symptom, or disease that occurs in a patient administered a medical treatment, whether the event is considered related or unrelated to the medical treatment. This study will utilize the Common Terminology Criteria for Adverse Events version 3.0 (CTCAE v3.0) for defining and grading specific adverse events. A copy of the CTCAE v3.0 can be downloaded from the CTEP home page at http://ctep.cancer.gov/reporting/ctc.html. A GOG CTCAE v3.0 Manual is also available on the GOG member web site (http://www.gog.org under MANUALS) and can be mailed to the institution registering a patient to this study if requested. 10.12 Reporting Expedited Adverse Events Depending on the phase of the study, use of investigational or commercial agents, and role of the pharmaceutical sponsor, an AE report may need to reach multiple destinations. For patients participating on a GOG trial, all expedited AE reports should be submitted by using the CTEP automated system for expedited reporting (AdEERS). All AdEERS submissions are reviewed by GOG before final submission to CTEP. Submitting a report through AdEERS serves as notification to GOG, and satisfies the GOG requirements for expedited AE reporting. All adverse reactions will be immediately directed to the Study Chair for further action. The requirement for timely reporting of AEs to the study sponsor is specified in the Statement of Investigator, Form FDA-1572. In signing the FDA-1572, the investigator assumes the responsibility for reporting AEs to the NCI. In compliance with FDA regulations, as contained in 21 CFR 312.64, AEs should be reported by the investigator. -54- GOG-0241 10.13 Phase 2 and 3 Trials Utilizing a Commercial Agent: AdEERS Expedited Reporting Requirements for Adverse Events That Occur Within 30 Days of the Last Dose of Any Commercial Study Agent Reporting Requirements for Adverse Events that occur within 30 Days¹ of the Last Dose of the Commercial Agent on Phase 2 and 3 Trials Grade 1 Grade 2 Grade 2 Grade 3 Unexpected Unexpected With Without and Expected Unexpected Expected Hospitali- Hospitalization zation Unrelated Unlikely Not Required Possible Probable Definite Not Required Not Required Not 7 Calendar Required Days 7 Calendar Not 7 Calendar Days Required Days Grade 3 Grades 4 & 52 Grades 4 & 52 Expected With Hospitalization Without Unexpected Expected Hospitalization Not Required 7 Calendar Days Not Required 7 7 Calendar Calendar Days Days 7 Calendar Days 7 Calendar Days Not Required 24-Hrs; 7 3 Calendar Calendar Days Days 1 Adverse events with attribution of possible, probable, or definite that occur greater than 30 days after the last dose of treatment with a commercial agent require reporting as follows: AdEERS 24-hour notification followed by complete report within 3 calendar days for: Grade 4 and Grade 5 unexpected events AdEERS 7 calendar day report: Grade 3 unexpected events with hospitalization or prolongation of hospitalization Grade 5 expected events 2 Although an AdEERS 24-hour notification is not required for death clearly related to progressive disease, a full report is required as outlined in the table. Please see exceptions below under the section entitled, “Additional Instructions or Exceptions to AdEERS Expedited Reporting Requirements for Phase 2 and 3 Trials Utilizing a Commercial Agent.” March 2005 Note: All deaths on study require both routine and expedited reporting regardless of causality. Attribution to treatment or other cause must be provided. Expedited AE reporting timelines defined: “24 hours; 3 calendar days” – The investigator must initially report the AE via AdEERS within 24 hours of learning of the event followed by a complete AdEERS report within 3 calendar days of the initial 24-hour report. “7 calendar days” – A complete AdEERS report on the AE must be submitted within 7 calendar days of the investigator learning of the event. Any medical event equivalent to CTCAE grade 3, 4, or 5 that precipitates hospitalization (or prolongation of existing hospitalization) must be reported regardless of attribution and designation as expected or unexpected with the exception of any events identified as protocolspecific expedited adverse event reporting exclusions. Any event that results in persistent or significant disabilities/incapacities, congenital anomalies, or birth defects must be reported to GOG via AdEERS if the event occurs following treatment with a commercial agent. -55 GOG-0241 Use the NCI protocol number and the protocol-specific patient ID provided during trial registration on all reports. Additional Instructions or Exceptions to AdEERS Expedited Reporting Requirements for Phase 2 and 3 Trials Utilizing a Commercial Agent: There are no additional instructions or exceptions to AdEERS expedited reporting requirements for this protocol. 10.14 Procedures for Expedited Adverse Event Reporting: 10.141 AdEERS Expedited Reports: Expedited reports are to be submitted using AdEERS available at http://ctep.cancer.gov. The NCI guidelines for expedited adverse event reporting requirements are also available at this site. Please consult these guidelines for secondary malignancy (including AML, MDS) reporting requirements. 10.15 Automated CDUS reporting For studies using commercial agents, the GOG Statistical and Data Center (SDC) routinely reports adverse events electronically to the CTEP Clinical Data Update System (CDUS Version 3.0). The SDC submits this data quarterly. The AEs reported through AdEERS will also be included with the quarterly CDUS data submissions. -5610.2 GOG-0241 GOG DATA MANAGEMENT FORMS The following forms must be completed and submitted to the GOG Statistical and Data Center (SDC) in accordance with the schedule below. All forms except the BDR Form and Pathology report must be submitted via the SDC Electronic Data Entry System (SEDES) which is available through the GOG website (www.gogstats.org). The BDR Form should be submitted via mail. The GOG Uploader Application in SEDES is an alternate method for submitting pathology reports and BDR to the GOG SDC. Form Due within Copies* Comments Weeks 2 Event Registration 2 Form OSO 2 Registration 2 Form MEDH 2 Registration 2 Form DR 4 Registration 2 Form BDR**** 4 Registration 2 Form D2M*** 4 Registration 2 Primary disease: Form F Pathology Report Slides 6 6 6 Registration Registration Registration 2 3 ** Submit to SDC via postal mail or via report uploader Submit to SDC via postal mail or via report uploader Form R Recurrent or Persistent Disease: Form F Pathology Report Slides Immunohistochemistry on section of primary tumor ***** Form D2R 6 6 6 Registration Registration Registration 2** 3** ** 12 2 N/A 2 Form D2M*** 2 Form T 2 Form Q0 2 Form Q 2 Form SP-0241-FP01 Form SP-0241-FP02 8 8 Registration Completion of each cycle of therapy Clinical response assessment Beginning of each subsequent cycle Completion of study Rx and change in Rx Disease progression; death; normal follow-up Registration Registration * ** Submit to SDC via postal mail or via report uploader 2 2 2 2 N/A N/A quarterly for 2 years, semiannually for 3 more years Online (SEDES) Online (SEDES) The number of required copies including the original form which must be sent to the Statistical and Data Center. At least one representative H&E stained slide(s) documenting the primary site, cell type and Stage of disease. For recurrent or persistent disease submit Form F, Pathology Report, and slides only if histologically documented. -57- GOG-0241 *** The original form is retained in the clinic and data is appended to this form each time the disease is reassessed. Three copies of the form are submitted to the Statistical and Data Center at baseline and each time clinical response is evaluated. **** This form is only used in studies with measurable disease along with DR form or in studies where the C form is being used. ***** In addition to the surgical pathology report and representative H&E stained slides, immunohistochemistry must be performed upon a section of the primary tumor with antibodies directed against CK7, CK 20, and CDX2, and the slides provided for review. (See Section 7.2) This study will be monitored by the Abbreviated/Complete Clinical Data System (CDUS) Version 3.0. CDUS data will be submitted quarterly to CTEP by electronic means. -5811.0 GOG-0241 STATISTICAL CONSIDERATIONS 11.1 Design Summary This is a randomized, 2x2 factorial (four-arm) Phase III clinical trial in patients with a histologic diagnosis of mucinous adenocarcinoma of the ovary or the fallopian tube with either optimal (≤ 1 cm residual disease) or suboptimal residual disease following initial surgery. The data from this trial will be combined with, and analyzed with, data from the mEOC-1 trial being run by Cancer Research UK: “A multicentre randomized trial comparing oxaliplatin + capecitabine versus carboplatin + paclitaxel in patients with previously untreated mucinous ovarian (mEOC) cancer.” All analyses will be based on the data from the combined GOG and GCIG trials. All patients (including both U.S. and non-U.S. patients) will be registered centrally at the GOG Statistical and Data Center. Prior to registration, eligibility will be reviewed via Fast Fact Sheet verification. All reports will include a complete accounting of all patients registered to this protocol. Patients will be randomized in a 1:1:1:1 ratio to receive one of the following treatment arms: Carboplatin + Paclitaxel Oxaliplatin + Capecitabine Carboplatin + Paclitaxel with Bevacizumab Oxaliplatin + Capecitabine with Bevacizumab The randomization will be stratified using using minimization, using the folllowing stratification factors: 1. Disease status: (i) No gross residual disease (i.e. residual disease = 0) (ii) Residual disease = >0 2. Stage: (i) Recurrent stage I disease (chemonaϊve) (ii) Not recurrent - stages II – IV 3. Country The assigned study regimen will not be revealed until after the patient has been successfully registered onto the study. All reports will include an accounting of all patients registered onto the study, regardless of their eligibility status or compliance to the assigned treatment. 11.2 Principal Parameters The principle parameters employed to evaluate the primary, secondary, and translational research objectives are: 11.21 Primary efficacy endpoint: overall survival 11.22 Secondary endpoints: -59- GOG-0241 11.221 Efficacy: progression-free survival and response rate 11.222 Safety: frequency and severity of adverse effects as assessed with CTC AE version 3.0 11.223 Quality of life: FACT-O TOI and FACT-GOG/NTX4 11.23 Translational Research Endpoints: IgM and IgG antibody titers by ELISA to antigens Tn-MUC1-32mer, GM2, Globo-H, TF, sTN, and Tn 11.3 Accrual Goal, Accrual Rate, and Study Duration The primary endpoint of trial is overall survival. The expected median survival in this patient population given carboplatin plus paclitaxel is about 12 months. The GCIG trial aims to detect increases in median survival of at least 5 months, from 12 to 17 months (or a hazard ratio of 0.71) for both capecitabine plus oxaliplatin versus carboplatin plus paclitaxel and for bevacizumab versus no bevacizumab. This treatment effect would be considered clinically worthwhile. We assume constant hazard ratios for each treatment effect, which would mean multiplicative effects and thus no interaction on the hazard ratio scale. Based on a recruitment period of 5 years and a follow-up period of 18 months after the end of recruitment, the GCIG trial requires 300 patients in the study in order to give the study 80% power for each main comparison. Allowing for a 10% dropout rate, the trial aims to recruit 332 in total. Two-sided tests with α=0.05 will be used. Table 11.1 shows the expected median survival in each treatment arm assuming a hazard ratio for 0.71 for each treatment main effect and assuming no interaction. Table 11.1: Expected Median Survival Time (months) by Treatment Group Carboplatin plus paclitaxel Capecitabine plus oxaliplatin No bevacizumab 12 17 Bevacizumab 17 24 Note: assuming a hazard ratio for 0.71 for each treatment main effect and no interaction. The GCIG protocol is projected to enroll the 332 patients in 5 years. In the GOG portion of the study, the projected accrual is 12 patients per year or a total of 60 patients across five years of accrual. This is based on the following: GOG-0182 accrued 59 patients who were classified as having mucinous cell type over a period of 43 months. Of these, 17 were excluded for the following reasons: second primary (5), wrong cell type (3), wrong primary (7), and inadequate pathology (2). Some of the excluded patients in GOG-0182 could have been eligible for this study, boosting the expected accrual rate. However, the rate of accrual to GOG-0182 was very high, so a comparable accrual rate for this study may be considered optimistic. Therefore, the projected accrual rate for this study is 12 patients per year. -6011.4 GOG-0241 Design and Analyses for the Primary Objective The primary outcome of overall survival will be examined using Kaplan-Meier curves, and comparisons between each set of two arms (‘oxaliplatin + capecitabine’ vs. ‘carboplatin + paclitaxel,’ and ‘bevacizumab’ vs. ‘no bevacizumab’) will be based on the logrank test and the hazard ratio (with 95% CI). The median survival time (with 95% CI) will also be calculated for each group. A stratified log rank test could be performed, based on the randomization strata and the other treatment randomization. For example, bevacizumab analysis will be stratified by residual disease, recurrent vs. non-recurrent and chemotherapy regimen. Cox regression will be used to assess the effects of the randomization strata (disease status and stage) and other prospectively defined risk factors on these two outcomes (such as age), and any interactions with the treatment group. The main effect size will be quantified by the hazard ratio and 95% CI. Multivariate regression will also be used to examine whether there is an interaction between oxaliplatin + capecitabine and bevacizumab. The power to evaluate interactions in the study is limited. Assuming the overall main effect hazard ratio of 0.71 is maintained with the underlying median survival times of 12 and 17 months in the treatment arms of interest (e.g., capecitabine plus oxaliplatin and carboplatin plus paclitaxel), Table 11.2 shows the resulting power for a variety of interactions. Table 11.2: Power to Detect Specified Interactions Factor Level† Power to Detect Specified Interaction – + Ratio of Hazard Ratios 0.91 0.49 2.02 80% 0.89 0.51 1.75 73% 0.83 0.55 1.50 49% † Factor levels could be, e.g., bevacizumab vs. no bevacizumab, or KRAS mutation (yes or no). Note: based on two-tailed test of interaction with α=0.10. To further help judge which treatment is best, methods for survival endpoints in factorial trials could be used, e.g., see Green S.(2006).56 All analyses will be performed on an intention-to-treat basis. 11.5 Interim Analysis A futility analysis will be performed after a third of the patients have been entered (n=110) and followed up for six months. Recruitment to the trial will continue during the 6-month follow-up. This will determine whether either of the experimental arms are unlikely to be associated with an improvement in survival. Formal futility rules will be developed in partnership with the Independent Data Monitoring Committee (IDMC) prior to reviewing the data. Methods like those described by Whitehead et al57 and statistical software such as PEST 4 (Reading University) can be used to estimate the probability of observing a hazard ratio of 0.71 or lower, if the trial continued to the end -61- GOG-0241 but given the data up to that point in time. A low probability of less than 10% could be used in support of stopping early. Because futility analyses are based on several assumptions and are, in effect, a forecast of data in the future, the futility analysis of overall survival alone will not be used to determine whether the trial stops early. We will also examine other outcome measures such as response rates and time to progression. This information, together with data on toxicity, will be used by the IDMC to decide whether oxaliplatin and capecitabine, or bevacizumab, is so ineffective that the trial or part of the trial (i.e. one of the randomizations) should discontinue. The interim analyses will also be used to examine whether there is likely to be an interaction between bevacizumab and chemotherapy regimen (carboplatin + paclitaxel vs. oxaliplatin + capecitabine), e.g. the survival using both regimens is greater than that expected by adding the survival when each regimen is used on its own. If there is evidence of an interaction (to be examined by the IDMC), the target sample size could be increased to increase the statistical power to examine it. In the absence of any current evidence on this, it is not possible to estimate how many more patients would be recruited, it may, for example, be 50-75 more in the bevacizumab arm. As part of ongoing quality assurance, investigators will assess what proportion of patients enrolled actually have mucinous ovarian cancer. At an interim analysis this data will be used to determine whether the sample size is appropriate and the study remains feasible. After the interim analysis has been conducted, if either comparison (ie bevacizumab vs. no bevacizumab, or carboplatin + paclitaxel vs oxaliplatin + capecitabine) is judged to be futile, that particular randomization will cease, and the trial would continue for the other randomization, i.e. as a two-arm study. 11.6 Trial Monitoring The data will be reviewed (approximately annually) by an Independent Data Monitoring Committee (IDMC), consisting of at least two clinicians not entering patients into the trial and an independent statistician. The IDMC will be asked to recommend whether the accumulated data from the trial, together with results from other relevant trials, justifies continuing recruitment of further patients. A decision to discontinue recruitment, in all patients or in selected subgroups will be made only if the result is likely to convince a broad range of clinicians including participants in the trial and the general clinical community. If a decision is made to continue, the IDMC will advise on the frequency of future reviews of the data on the basis of accrual and event rates. The IDMC will make confidential recommendations to the trial steering committee (TSC). The role of the TSC is to act on behalf of the funder, to provide overall supervision for the trial, to ensure that it is conducted in accordance with ICH GCP, and to provide advice through its independent Chairman. This independent committee will review the recommendations from the IDMC and will decide on continuing or stopping the trial, or modifying the protocol. -62- GOG-0241 The GOG Data Safety and Monitoring Board (GOG-DSMB) reviews accumulating summaries of toxicities and all serious adverse event (SAE) reports on an ongoing basis (not efficacy results). This committee also reviews those deaths in which study treatment may have been a contributing cause. The GOG-DSMB reports to the GOG Data Monitoring Committee (GOG-DMC) and it may recommend study amendments pertaining to patient safety. Data sheets from patients on this protocol will be reviewed before each semi-annual meeting and will also be reviewed by the Study Chairperson in conjunction with the Statistical and Data Center. In some instances, because of unexpectedly severe toxicity, the Statistical and Data Center may elect, after consultation with the Study Chairperson and the Medical Oncology Committee, to recommend early suspension of a study. The frequency and severity of all toxicities are tabulated from submitted case report forms and summarized for review by the study chairperson, Ovarian Committee, and GOG-DSMB in conjunction with each semi-annual GOG meeting. For studies sponsored by the Cancer Therapy Evaluation Program (CTEP) of the National Cancer Institute (NCI), standardized toxicity reports are also submitted to the drug and disease monitors at the Investigational Drug Branch (IDB) and Clinical Investigation Branch (CIB). 11.7 Analyses for Secondary Objectives PFS will analyzed using the same methods described above the analysis of overall survival. The comparison of response rates (proportion of patients with complete and partial response, stable and progressive disease) between treatment groups, will use a chisquare test. The difference in response rates between the groups and the odds ratio (with corresponding 95% C.I.s) will also be calculated. Toxicity grades will be tabulated showing the maximum toxicity grade experienced by each patient. The proportions of patients experiencing a maximum grade of 3 or above will be compared between the treatment groups. Analysis of the QOL data will use the observed scores at each time point, where available. For simplicity, we will examine the change from baseline to each of time points specified in section 6.2 and 6.3. However, the main analysis will be a repeated measures analysis (for example, based on a mixed model, Proc Mixed in SAS) used to simultaneously compare all QOL scores between the two treatment groups. This method allows for (i) that each patient provides several QOL scores, (ii) that scores withinpatients are likely to be correlated, and (iii) that QOL scores between proximate visits are likely to be more correlated than separated visits. All tests for exploratory and translational research objectives will be done using onesided tests with α=0.05. All analyses will be performed on an intention-to-treat basis. -6311.8 GOG-0241 Analyses for Exploratory and Translational Research Objectives The numbers and percentages of patients with expression of VEGF and EGF ligand, receptor, and activated receptor will be tabulated, as will the number and percentage of patients with mutations in the KRAS oncogene. Interactions between the mutational status of the KRAS oncogene and treatment will be examined. The power to test these interactions are addressed above in section 11.4. 11.9 Anticipated Gender and Minority Inclusion This study restricts entry to women by nature of the site of the disease. The table below lists the projected percentages in the GOG portion of the study of patients by race (based on GOG protocol 0182). There are no data that support differences in the intervention effects between racial/ethnic subgroups; therefore, the study design does not involve race. However, subsets defined by white and non-white will be analyzed in this study to investigate this important question with the current therapies. The trial is registered at clinicaltrials.gov, and all patients who meet eligibility criteria are invited to participate. 2% Asian 7% Black 2% Other (e.g., American Indian/Alaskan Native) 89% White 100% Total -64- GOG-0241 12.0 BIBLIOGRAPHY 1. McGuire V, Jesser CA, Whittemore AS. Survival among U.S. women with invasive epithelial ovarian cancer. Gynecol Oncol 2002;84:399-403. 2. Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2006. CA Cancer J Clin 2006 MarApr;56:106-130. 3. Sherman ME, Mink PJ, Curtis R, et al. Survival among women with borderline ovarian tumors and ovarian carcinoma: a population-based analysis. Cancer 2004;100:1045-1052. 4. Hess V, A'Hern R, Nasiri N, et al. Mucinous epithelial ovarian cancer: a separate entity requiring specific treatment. J Clin Oncol 2004;22:1040-1044. 5. Marquez RT, Baggerly KA, Patterson AP, et al. Patterns of Gene expression in different histotypes of epithelial ovarian cancer correlate with those in normal fallopian tube, endometrium, and colon. Clin Cancer Res 2005; 11 (17): 6116-6216. 6. Gemignani ML, Schlaerth AC, Bogomolniy F, et al. Role of KRAS and BRAF gene mutations in mucinous ovarian carcinoma. Gynecol Oncol 2003;90:378-381. 7. Boguski, M. S., and McCormick, F. (1993) Science 366, 643-654 8. Williams AC, Browne SJ, Yeudal WA, et al. Molecular events including p53 and k-ras alterations in the in vitro progression of a human colorectal adenoma cell line to an adenocarcinoma. Oncogene 1993;8:3063-3072. 9. Muggia FM, Braly PS, Brady MF, et al. Phase III randomized study of cisplatin versus paclitaxel versus cisplatin and paclitaxel in patients with suboptimal stage III or IV ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol 2000;18:106-115. 10. McGuire WP, Hoskins WJ, Brady MF, et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med 1996;334:1-6. 11. Piccart MJ, Bertelsen K, James K, et al. Randomized intergroup trial of cisplatin-paclitaxel versus cisplatin-cyclophosphamide in women with advanced epithelial ovarian cancer: threeyear results. J Natl Cancer Inst 2000;92:699-708. 12. de Gramont A, Figer A, Seymour M, et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol 2000;18:29382947. 13. Saltz LB, Douillard JY, Pirotta N, et al. Irinotecan plus fluorouracil/leucovorin for metastatic colorectal cancer: a new survival standard. Oncologist 2001;6:81-91. 14. Goldberg RM, Sargent DJ, Morton RF, et al. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J Clin Oncol 2004;22:23-30. -65- GOG-0241 15. Scheithauer W, Kornek GV, Raderer M, et al. Intermittent weekly high-dose capecitabine in combination with oxaliplatin: a phase I/II study in first-line treatment of patients with advanced colorectal cancer. Ann Oncol 2002;13:1583-1589. 16. Borner MM, Dietrich D, Stupp R, et al. Phase II study of capecitabine and oxaliplatin in firstand second-line treatment of advanced or metastatic colorectal cancer. J Clin Oncol 2002;20:1759-1766. 17. Fracasso PM, Blessing JA, Morgan MA, et al. Phase II study of oxaliplatin in platinumresistant and refractory ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol 2003;21:2856-2859. 18. Liavaag AH, Dorum A, Fossa SD, Trope C, Dahl AA. Controlled study of fatigue, quality of life, and somatic and mental morbidity in epithelial ovarian cancer survivors: how lucky are the lucky ones? J Clin Oncol 2007;25: 2049-56. 19. Sun CC, Ramirez PT, Bodurka DC. Quality of life for patients with epithelial ovarian cancer. Nat Clin Pract Oncol 2007;4: 18-29. 20. Wenzel LB, Donnelly JP, Fowler JM, Habbal R, Taylor TH, Aziz N, Cella D. Resilience, reflection, and residual stress in ovarian cancer survivorship: a gynecologic oncology group study. Psychooncology 2002;11: 142-53. 21. Goldberg RM. Advances in the treatment of metastatic colorectal cancer. Oncologist 2005;10 Suppl 3: 40-8. 22. Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, Erdkamp FL, Vos AH, van Groeningen CJ, Sinnige HA, Richel DJ, Voest EE, Dijkstra JR, Vink-Borger ME, Antonini NF, Mol L, van Krieken JH, Dalesio O, Punt CJ. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med 2009;360: 563-72. 23. Bookman MA, Brady MF, McGuire WP, Harper PG, Alberts DS, Friedlander M, Colombo N, Fowler JM, Argenta PA, De Geest K, Mutch DG, Burger RA, Swart AM, Trimble EL, AccarioWinslow C, Roth LM. Evaluation of New Platinum-Based Treatment Regimens in AdvancedStage Ovarian Cancer: A Phase III Trial of the Gynecologic Cancer InterGroup. J Clin Oncol 2009. 24. Borner MM, Dietrich D, Stupp R, Morant R, Honegger H, Wernli M, Herrmann R, Pestalozzi BC, Saletti P, Hanselmann S, Muller S, Brauchli P, Castiglione-Gertsch M, Goldhirsch A, Roth AD. Phase II study of capecitabine and oxaliplatin in first- and second-line treatment of advanced or metastatic colorectal cancer. J Clin Oncol 2002;20: 1759-66. 25. Argyriou AA, Polychronopoulos P, Iconomou G, Chroni Bookman MA, Brady MF, McGuire WP, Harper PG, Alberts DS, Friedlander M, Colombo N, Fowler JM, Argenta PA, De Geest K, Mutch DG, Burger RA, Swart AM, Trimble EL, Accario-Winslow C, Roth LM. Evaluation of New Platinum-Based Treatment Regimens in Advanced-Stage Ovarian Cancer: A Phase III Trial of the Gynecologic Cancer InterGroup. J Clin Oncol 2009. -66- GOG-0241 E, Kalofonos HP. A review on oxaliplatin-induced peripheral nerve damage. Cancer Treat Rev 2008;34: 368-77. 26. Basen-Engquist K, Bodurka-Bevers D, Fitzgerald MA, Webster K, Cella D, Hu S et al. Reliability and validity of the functional assessment of cancer therapy-ovarian. J Clin Oncol 19:1809-17, 2001. 27. Cain JM, Wenzel LB, Monk BJ, Cella D. Palliative care and quality of life considerations in the management of ovarian cancer. In: Gershenson DM, McGuire WP. Ovarian Cancer: Controversies in Management. New York: Churchill Livingstone 281-307, 1998. 28. Huang HQ, Brady, MF, Cella D, Fleming G. Validation and reduction of FACT/GOG-Ntx subscale for platinum/paclitaxel-induced neurologic symptoms: a gynecologic oncology group study. International J of Gyn Cancer, in press. 29. Kabbinavar F, Hurwitz HI, Fehrenbacher L, et al. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J Clin Oncol 2003;21:60-65. 30. Saltz LB, Clarke S, Diaz-Rubio E, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008; 26(12):2013-9. 31. Cooper BC, Ritchie JM, Broghammer CL, et al. Preoperative serum vascular endothelial growth factor levels: significance in ovarian cancer. Clin Cancer Res 2002;8:3193-3197. 32. Abu-Jawdeh GM, Faix JD, Niloff J, et al. Strong expression of vascular permeability factor (vascular endothelial growth factor) and its receptors in ovarian borderline and malignant neoplasms. Lab Invest 1996;74:1105-1115. 33. Burger RA, Sill MW, Monk BJ, et al. Phase II trial of bevacizumab in persistent or recurrent epithelial ovarian cancer or primary peritoneal cancer: A Gynecologic Oncology Group study. J Clin Oncol 2007; 25:5165-5171. 34. Cannistra SA, Matulonis UA, Penson RT, et al. Phase II study of bevacizumab in patients with platinum-resistant ovarian cancer or peritoneal serous cancer. J Clin Oncol 2007; 25:51805186. 35. Garcia AA, Hirte H, Fleming G, et al. Phase II clinical trial of bevacizumab and low-dose metronomic oral cyclophosphamide in recurrent ovarian cancer: A trial of the California, Chicago, and Princess Margaret Hospital Phase II Consortia. J Clin Oncol 2008; 26:76-82. 36. Spannuth et al, International Journal of Cancer, 124:1045-1053, 2009 -67- GOG-0241 37. Berchuck A, Rodriguez GC, Kamel A, et al. Epidermal growth factor receptor expression in normal ovarian epithelium and ovarian cancer. I. Correlation of receptor expression with prognostic factors in patients with ovarian cancer. Am J Obstet Gynecol 1991;164:669-674. 38. Niikura H, Sasano H, Sato S, Yajima A. Expression of epidermal growth factor-related proteins and epidermal growth factor receptor in common epithelial ovarian tumors. Int J Gynecol Pathol 1997;16:60-68. 39. Gemignani M, Schlaerth AC, Bogomolniy F, et al. Role of KRAS and BRAF gene mutations in mucinous ovarian carcinomas. Gynecol Oncol 2003;90:378-381. 40. Kinzler KW, Vogelstein B. Colorectal tumors. In: Vogelstein B, Kinzler KW, eds. The Genetic Basis of Human Cancer. New York: McGraw-Hill, 1998:565-590. 41. Santesson, L. and H. Kottmeier (1968). General classification of ovarian tumors. UICC Monograph Series. F. Gentil and A. Junqueira. New York, Springer-Verlag. 11: 1-8. 42. Hart, W. and H. Norris (1973). "Borderline and malignant mucinous tumors of the ovary." Cancer 31: 1031-1045. 43. Chaitin, B., D. Gershenson, et al. (1985). "Mucinous tumors of the ovary." Cancer 55: 19581962. 44. Watkin, W., E. Silva, et al. (1992). "Mucinous carcinoma of the ovary: Pathologic prognostic factors." Cancer 69: 208-212. 45. Hoerl, H. and W. Hart (1998). "Primary ovarian mucinous cystadenocarcinomas." Am J Surg Pathol 22: 1449-1462. 46. Rodriguez, I. and J. Prat (2002). "Mucinous tumors of the ovary: A clinicopathology analysis of 75 borderline tumors (of intestinal type) and carcinomas." Am J Surg Pathol 26: 139-152. 47. Hart, W. (2004). "Mucinous tumors of the ovary: a review." Int J Gynecol Pathol 24: 4-24. 48. Lee, K. and R. Young (2003). "the distinction between primary and metastatic mucinous carcinomas of the ovary." Am J Surg Pathol 27: 281-292. 49. Riopel, M., B. Ronnett, et al. (1999). "Evaluation of diagnotstic criteria and behavior of ovarian intestinal-type mucinous tumors." Am J Surg Pathol 23: 617-35. 50. Holtz, F. and W. Hart (1982). "Kruckenberg tumors of the ovary." Cancer 50: 2438-2447. 51. Seidman, J., R. Kurman, et al. (2003). "Primary and metastatic mucinous adenocarcinomas in the ovaries." Am J Surg Pathol 27: 985-993. 52. DeLott, L., C. Morrison, et al. (2005). "CDX2 is a useful marker of intestinal-type differentiation." Arch Pathol Lab Med 129: 1100-1105. -68- GOG-0241 53. Logani, S., E. Oliva, et al. (2005). "Use of novel immunohistochemical markers expressed in colonic adenocarcinoma to distinguish primary ovarian turmos from metastatic colorectal adenocarcinoma." Modern Pathology 18: 19-25. 54. Gamelin L, Boisdron-Celle M, Delva R, Guérin-Meyer V, Ifrah N, Morel A, and Gamelin E. Prevention of Oxaliplatin-Related Neurotoxicity by Calcium and Magnesium Infusions. Clin Cancer Res 2004;10:4055-4061 55. Tooze JA, Grunwald GK, Jones RH. Analysis of repeated measures data with clumping at zero. Statistical Methods in Medical Research 2002;11:341-355. 56. Factorial designs with time to event endpoints. Handbook of Statistics in Clinical Oncology, 2006, 2nd ed. J Crowley CRC Press: 181 – 189. 57. Whitehead J, Matsushita T. Stopping clinical trials because of treatment ineffectiveness: a comparison of a futility design with a method of stochastic curtailment. Statistics in Medicine, 2003; 22(5):677-687