Methods - Nature
advertisement

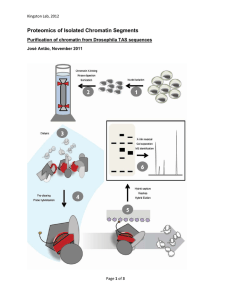
Methods Purification of 15-lipoxygenase (15-LOX) 15-LOX was purified from rabbit reticulocytes. The lysate was prepared according to Jackson and Hunt1. After sedimentation of the ribosomes, the supernatant was precipitated with 66% ammonium sulphate. The resuspended pellet was dialyzed against 50 mM KOAc, 50 mM Hepes pH7.5, 5 mM Mg(OAc)2 and passed through a Q Sepharose column. The flow-through was precipitated with ammonium sulphate, dialyzed against 25 mM KOAc, 10 mM Hepes pH7.5, 5 mM Mg(OAc)2 and applied to an S Sepharose column. The LOX-containing fractions, eluting around 130 mM potassium acetate, were pooled and concentrated by ammonium sulphate precipitation and dialysis against 50 mM KOAc, 50 mM Hepes pH7.5, 5 mM Mg(OAc)2. After this step, the enzyme is more than 95% pure as judged by Coomassie staining. To determine the identity of the purified enzyme, 15-LOX was transferred from an SDS gel to nitrocellulose, digested with trypsin, and the resulting peptides were analyzed by mass spectrometry2. Antibodies We have raised peptide specific antibodies in rabbits against the C-terminal amino acid sequence of 15-LOX (YLRPSIVENSVAI) and used this peptide to affinity purify the antibodies. Isolation of organelles, binding and release assays, and biochemical characterization The organelles used in this study were purified from rat liver by standard protocols. rER and sER were a gift from G. Kreibich, Golgi membranes from C. Hughes, peroxisomes and mitochondria were provided by W. W. Just. To test the association of 15-LOX with membranes, we have used sedimentation assays. Organelles (final concentration approximately 1 mg of protein/ml) were incubated with 1 mg/ml 15LOX or buffer for 5 minutes at 26 ˚C and then for ca. 10 minutes on ice in a volume of 40 µl containing 50 mM Hepes/KOH pH 7.5, 50 mM KOAc, 5 mM Mg(OAc) 2, and 1 mM DTT. The reaction mixture was then layered on top of a 160 µl cushion consisting of 500 mM sucrose in incubation buffer and centrifuged for 10 minutes in a TL 100 rotor (Beckman) at 100000 rpm. The pellets were resuspended in SDSsample buffer and separated by SDS-PAGE and the proteins visualized by Coomassie Blue staining or Western blotting with ECL detection (Amersham). For the release experiment, a fresh mitochondrial preparation from bovine liver was used. Incubation conditions were as described for the binding experiment, but with an incubation time of 2 hours. Aliquots of supernatant and pellet fractions after centrifugation were probed with antibodies to lumenal and integral membrane proteins of the respective organelle provided by W. W. Just (catalase and PMP69), W. A. Fenton (methylmalonyl-CoA mutase), M. F. Bauer (Tim23), G. Kreibich (Ribophorins I and II), M. Gmachl (GOS28) Stressgen (PDI, protein disulphide isomerase) and Sigma (albumin). Inside-out ghost membranes were prepared from fresh rabbit blood according to Steck and Kant3, rER membranes and EDTA-high salt stripped rough microsomes (EKRM) from dog pancreas according to Walter and Blobel 4. Inhibition of 15-LOX was achieved by pretreating the enzyme for 5 minutes at 26 ˚C with 200 µM npropyl gallate prior to incubation with rER membranes or inside-out ghosts. Carbonate extraction of rER membranes was carried out according to Fujiki et al. 5, phase separation in Triton X-114 according to Bordier6. Liposome preparation and electron microscopy Multilamellar vesicles containing carboxyfluorescein were generated from either 1Stearoyl-2-Arachidonyl-sn-Glycero-3-Phosphocholine or 1-Oleyl-2-Palmitoyl-snGlycero-3-Phosphocholine (Avanti Polar Lipids) 7. Briefly, 20 mg of phospholipid were dissolved in chloroform/methanol (2:1) and the solvent was evaporated to dryness in a round glass flask on a rotary evaporator. 2 ml buffer containing 1 mM carboxyfluorescein (Molecular Probes) were added along with glass beads, and the phospholipid was resuspended by gently shaking. After purification by gel filtration on Sephadex G-25 (1 x 25 cm; Pharmacia), the corresponding liposomes were incubated with either buffer or aprotinin (as control) or with 15-LOX or 1% Triton X-100 (to achieve 100% release of carboxyfluorescein). After resedimentation of the liposomes for 20 minutes at 45000 rpm in a TLA 45 rotor (Beckman), which is sufficient to sediment liposomes incubated with 15-LOX (not shown), all supernatants were adjusted to 1% Triton X-100 and fluorescence emission was measured at 515 nm (excitation wavelength 490 nm) with a Spex Fluorolog-2 spectrofluorometer. For electron microscopy, unilamellar vesicles were generated according to Paul et al.8 by solubilization of multilamellar liposomes (10 mg/ml) in buffer containing 5% octyl-polyoxyethylene (Bachem, Heidelberg) at a final concentration of 0.5 mg lipid/ml and dialysis against 20 mM Hepes/KOH pH7.5, 50 mM KOAc, 5 mM Mg(OAc)2, 1 mM DTT, 3 mM NaN3 for 72 hours at 30 ˚C. These vesicles were incubated with buffer or 0.85 mg/ml 15-LOX for 30 minutes at 26 ˚C, sedimented for 20 minutes at 14000 rpm in an Eppendorf centrifuge and resuspended in buffer. Samples for electron microscopy were washed with double distilled water on the grids prior to staining with 2% uranyl acetate. Micrographs were taken in a Philips EM 420 electron microscope. Lens Immunohistochemistry. Adult mouse eyes were fixed in 4% paraformaldehyde overnight, paraffin-embedded and sectioned horizontally at 5 µm. The sections were incubated in 50 mM NH4Cl in Phosphate-buffered saline (PBS) to quench autofluorescence, permeabilized with 0.075% saponin in PBS, and non-specific staining was blocked with 5% nonimmune goat serum, 0.2% bovine serum albumin in PBS. The primary affinitypurified anti-lipoxygenase rabbit antibodies was used at a 1:400 dilution and the mouse monoclonal antiserum produced against the C-terminus of rat Grp78/BiP (StressGen) was used at 1:200. Incubations were at room temperature for 2 hours. Indocarbocyanine (cy3)-conjugated goat anti-rabbit and Fluorescein Isothiocyanate (FITC)-conjugated goat anti-mouse secondary antibodies (Jackson ImmunoResearch) were used at 1:400 and 1:200 dilutions, respectively. The sections were coverslipped using Vectashield (Vector) and analyzed using epifluorescence illumination on an Axiovert 35 Zeiss microscope. Photographs were scanned and superposed using Adobe Photoshop. References 1. Jackson, R.J. & Hunt, T. Preparation and use of nuclease-treated rabbit reticulocyte lysates for the translation of eukaryotic messenger RNA. Methods Enzymol 96, 50-74 (1983). 2. Lui, M., Tempst, P. & Erdjument, B.H. Methodical analysis of protein- nitrocellulose interactions to design a refined digestion protocol. Anal Biochem 241, 156-66 (1996). 3. Steck, T.L. & Kant, J.A. Preparation of impermeable ghosts and inside-out vesicles from human erythrocyte membranes. Methods Enzymol 31A, 172-180 (1974). 4. Walter, P. & Blobel, G. Preparation of microsomal membranes for cotranslational protein translocation. Methods Enzymol 96, 84-93 (1983). 5. Fujiki, Y., Hubbard, A.L., Fowler, S. & Lazarow, P.B. Isolation of intracellular membranes by means of sodium carbonate treatment. J. Cell Biol. 93, 97-102 (1982). 6. Bordier, C. Phase Separation of Integral Membrane Proteins in Triton X-114 Solution. J. Biol. Chem. 256, 1604-1607 (1981). 7. New, R.R.C. Liposomes: a practical approach, pp. 37-39 (Oxford University Press, New York, 1990). 8. Paul, A., Engelhardt, H., Jakubowski, U. & Baumeister, W. Two- dimensional crystallization of a bacterial surface protein on lipid vesicles under controlled conditions. Biophys. J. 61, 172-188 (1992).