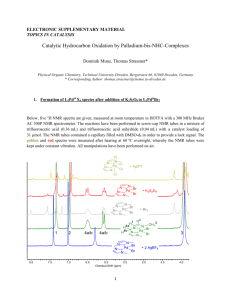
View/Open - Sacramento
advertisement

1 Chapter 1 INTRODUCTION Background The synthesis of heterocycles is of great importance in pharmaceutical and medicinal chemistry. The ever-increasing demand for novel biologically-active compounds and the laborious process of lead discovery and optimization have resulted in the continuous search for simple and efficient methods for generating libraries for biological screening. 1,2,3-Triazoles have not been isolated in any naturally occurring compounds,1 however the 1,2,3-triazole moiety has been utilized in many applications ranging from industrial to pharmaceutical uses. The applications of 1,2,3-triazoles are widespread, making 1,2,3-triazole derivatives a highly studied class of molecules. Triazoles belong to a class of compounds called azoles. An azole contains a fivemembered aromatic ring with at least one nitrogen atom and another heteroatom such as a nitrogen, sulfur, or oxygen. A 1,2,3-traizole structure contains three adjacent nitrogen atoms with three available substitution sites found at positions 1, 4 and 5 (Figure 2). Substitution Positions H N 2 N 1 5 N 3 4 Figure 2. Structure of an unsubstituted 1H-1,2,3-triazole. 2 Although the nitrogen at the N1 position is shown with a proton, this atom does not remain stationary. At both equilibrium and room temperature, the 1,2,3-triazole contains both 1H and 2H tautomers in dilute solutions. In more concentrated solutions and at lower temperatures, the 1H structure predominates.2-6 The tautomerizism at room temperature can be seen in the 1H NMR spectrum of dimethyl 1H-1,2,3-triazole-4,5-dicarboxylate (Figure 3). The observed peak for the N-H (A) is not a single sharp peak, but rather a broad peak 15.6 to 16.8 ppm due to the averaging of the triazole tautomer signals. The two different methyl esters (B) appear to be identical since an equilibrium between the 1H and 2H tautomers is established so rapidly, that at SpinWorks 2.3: room temperature, only a single signal appears in the NMR.7 It should be noted that if the proton is substituted with a larger group (i.e. phenyl), the larger group remains stationary and no tautomerization occurs. A H N B H3CO2C A B H3CO2C N N H N B N H3CO2C A B N H3CO2C x 256.000 16.4 PPM 16.0 15.0 16.0 14.0 B 15.6 13.0 12.0 11.0 10.0 9.0 8.0 7.0 6.0 file: C:\DocumentsandSettings\Cat Roush\My Documents\Under GradClass\CHEM189&198\Paper PublicationData\Dr KNMRspectraanddata\1H_diester inDMSO\1\fid expt: freq. <zg30> of 0ppm: 300.142603MHz transmitter freq.: 300.144690MHz processedsize: 32768complexpoints timedomainsize: 65536points LB: 0.000 GB: 0.0000 width: 6172.84Hz=20.566213ppm=0.094190Hz/pt number of scans: 16 5.0 4.0 3.0 2.0 1.0 0.0 Figure 3. 1H NMR Spectrum of dimethyl 1H-1,2,3-triazole-4,5-dicarboxylate in d6-DMSO. -1.0 3 The development of 1,2,3-triazoles for drug discovery and industrial use has been shown to be very versatile. The uses for triazoles have been found in various areas and are continuously growing. The applications of these triazoles are increasingly found in all aspects of drug discovery, ranging from cutting edge research through combinatorial chemistry and targettemplated in situ chemistry, to proteomics and DNA research using bioconjugation reactions. 8 These triazole products are more than just passive linkers; they readily associate with biological targets, through hydrogen bonding and dipole interactions.8 Derivatives of 1,2,3-triazole have been found to have anti-HIV,9 anti-allergenic,10 antimicrobial, cytostatic, virostatic, antiinflamatory11 and anti-bacterial12 activities. Triazoles are also being studied for the treatment of obesity13 and osteoarthritis.14 The increased interest in the 1,2,3-triazole is due to it being nontoxic, benign and stable. Triazoles are particularly interesting for medicinal use because they are more likely to be water soluble than normal aromatic compounds, and are stable in biological systems.15 On the industrial side, 1,2,3-triazoles are found in hydraulic fluids, agrochemicals (fungicides), and photochemical products.16, 17 They have also been used as herbicides, light stabilizers, fluorescent whiteners, optical brightening agents, pigments and corrosion retardants. 11, 18-20 This allows for the applications of 1,2,3-triazoles to grow exponentially due to their reliability, tolerance to a wide variety of functional groups, regiospecificity and the readily available starting materials. Through this, 1,2,3-triazoles are very attractive to use and apply in many fields. 4 1,3-Dipolar Cycloaddition Most of the classic reactions for the synthesis of heterocyclic are accomplished by cyclization, since cycloaddition reactions provide routes to heterocycles with well-defined substitution patterns.21 This can vary in complexity from a one-step synthesis using a single reaction component, to a multicomponent procedure with a large number of steps.20 The synthesis of the 1,2,3-triazole ring structure is typically accomplished by a 1,3-dipolar cycloaddition using an alkyne and an azide (Scheme 1). R2 N 1 C C R N N 1 R N 1 C R N R2 N C R1 Scheme 1. Alkyne and azide react to produce a 1,2,3-traizole. The approach of 1,3-dipolar cycloadditions constitutes a powerful tool in the synthesis of five-membered heterocyclic rings. This technique does not restrict what compounds may be synthesized. The Woodward-Hoffmann theories of orbital symmetry conservation, as well as frontier orbital theory have provided a basis for the understanding of these mechanisms and for interpreting the effect of substituents on the rates and selectivities of cycloadditions. 21, 22 1,3- Dipolar cycloadditions are an excellent method for constructing five membered rings with a wide variety of 1,3-dipoles commercially available today. 5 The concept of 1,3-dipolar cycloaddition originated when Rolf Huisgen published a review article in 1955.23 Huisgen discussed the resonance structures of CH2N2 which explained the valence bond description of 1,3-dipolar cycloaddition (Scheme 2). His breakthrough consisted of the realization that the description of diazoalkanes can be applied to a series of other structures (i.e. alkynes and azides) in which carbon, nitrogen, and oxygen are affected in the resonance structure that may hold a charge. HC N H2C N H2C N N N N Scheme 2. Resonance structures of CH2N2. The 1,3-dipolar cycloaddition, also known as the Huisgen cycloaddition, is a classic approach in organic chemistry consisting of the reaction of a dipolarophile with a 1,3-dipolar compound that allows for the production of various five-membered heterocycles.24 Most of dipolarophiles are alkenes, alkynes and molecules possessing related heteroatom functional groups (such as carbonyls and nitriles). A 1,3-dipole is a three-atom conjugated system with four π-electrons delocalized over the three atoms. The name 1,3-dipole was coined because it is impossible to write electron-paired resonance structures for these species without incorporating charges.25 1,3-dipolar compounds contain one or more heteroatoms and can be described as 6 having at least one resonance structure that represents a charged dipole. It is important to note that 1,3-dipolar species contain a heteroatom as the central atom. This allows the 1,3-dipolar species to be formally sp- or sp2-hybridized depending on whether or not there is a double bond orthogonal to the delocalized π-system. The 1,3-dipoles can be divided into two different types: 1) the allyl anion type such as nitrones, azomethine ylides which contains a nitrogen atom in the middle of the dipole, carbonyl ylides, or carbonyl imines, which contains an oxygen atom in the middle of the dipole and 2) the linear propargyl/allenyl anion type such as nitrile oxides, nitrilimines, nitrile ylides, diazoalkanes, or azides. These two types of 1,3-dipole are shown below in Table 1. 7 Table 1. 1,3-Dipoles useful in cycloaddition reactions.25 X Y Y Z X Linear Propargyl/Allyl Type N N Allyl Anion Type N N azides N N C HC HC HC N N O diazo C N C compounds HC Z N nitrones azomethine imides O N nitrile oxides C nitrile imides C nitrile sulfides C C azomethine ylides N N N O C carbonyl ylides S C nitrile ylides S C thiocarbonyl ylides 8 The substrates shown in Table 1 are examples of compounds that may be used in a 1,3dipolar cycloaddition reaction. These particular substrates are known as 1,3-dipoles. Dipolarophiles are alkenes, alkynes or molecules that possess a heteroatom functional group. The two π-electrons supplied by the dipolarophile and the four electrons of the dipolar compound participate in a concerted, pericyclic shift. The addition is stereoconservative (suprafacial), and the reaction is therefore a [2s+4s] cycloaddition, more commonly known as a 1,3-dipolar cycloaddition (Scheme 3).26, 27 The regioselectivity of the reaction depends on electronic and steric effects. Meaning, if there are functional groups on each of the substrates that are large, it is possible to obtain a single isomer. If not, then isomers will be formed and no regioselectivity will be observed. R2 R1 C C 1 R 2 pi N R1 N R2 N N N C C N R1 N 4 pi 1 C R N R2 N C R1 2s + 4s cycloaddition Scheme 3. Example of a 1,3-dipolar cycloaddition reaction. These five-membered heterocyclic rings are not limited to carbon, nitrogen, and oxygencontaining compounds, but also allow for phosphorus and sulfur to be included in the ring. There are various 1,3-dipoles (Table 1) that may be used to construct different 1,2,3-triazoles. The result is an ever-growing number of heterocyclic compounds that may be constructed using this ingeniously simple technique. 9 The 1,3-dipolar cyloaddition can be accomplished using an alkyne and an azide to produce a wide variety of 1,2,3-triazoles with great ease. This simple method allows for 1,2,3triazoles to be synthesized with only two components, although the approach does potentially lead to regioisomeric products. Sharpless and coworkers introduced the term “click chemistry” for this approach, which denotes the development of a set of powerful, highly reliable, and selective reactions for the rapid synthesis of useful new compounds and combinatorial libraries through heteroatom links. Click chemistry does not replace existing methods for drug discovery, but rather, it complements and extends them. It works well in conjunction with structure-based design and combinatorial chemistry techniques, and, through the choice of appropriate building blocks, can provide derivatives or mimics of ‘traditional’ pharmacophores, drugs and natural products.28, 29 The benefits of 1,3-dipolar cycloaddition allows for many new compounds to be synthesized quickly and efficiently. An increase in demand for fast and effective reactions makes the 1,3-dipolar cycloaddition reaction indispensible. Even though this technique is used frequently for many applications today, 1,2,3-triazoles were synthesized quite differently in the past. Many of the old synthesis techniques required harsh conditions, high temperatures, high pressures and long reaction times. Synthesizing 1,2,3-triazoles was a multi-step process which required purification for every step. The discovery of simpler and more straightforward techniques in synthesizing 1,2,3-triazoles would lead to the creation for the concept of 1,3-dipolar cycloaddition reactions. 10 Brief History for the Discovery and Optimization of the 1,2,3-Triazole The history of the 1,2,3-triazoles began with several isolated events that did not suggest to the chemists at the time that these compounds would produce any significant contribution to organic chemistry. In 1860, Zinin30 was investigating two isomeric compounds that were isolated from the nitration of diphenyldiazene-1-oxide and were identified as nitroazoxybenzene and isonitrosazoxybenzene (Scheme 4). When both isomers were reduced with ammonium sulfide, Zinin stated that isonitroazoxybenzene produced six equivilants of sulfur while nitroazoxybenzene only produced four equivalants. Since Zinin was using the older atomic weight (O = 8), it was not possible for him to determine a molecular formula of isonitroazoxybenzene that would have enabled him to make a successful identification of the nature of his compound. Later, in 1899, it was shown that Zinin’s reduction product was actually 2-phenylbenzotriazole-1-oxide, a derivative of the 1,2,3-triazole. O N N HNO3 N H2SO4 diphenyldiazene-1-oxide O O N N N O N + N O nitroazoxybenzene OH isonitrosazoxybenzene (NH4)2S O N N N 2-phenylbenzotriazole-1-oxide Scheme 4. The nitration of diphenyldiazene-1-oxide.30 11 Prior to 1888, all reactions that produced a 1,2,3-triazole were derivatives of benzotriazole. The discovery of simple, monocylic 1,2,3-triazoles is credited to Han von Pechmann. In 1888, von Pechmann synthesized a monocyclic triazole and correctly formulated the 1,2,3-triazole ring. He was able to do this for both 1H-1,2,3-triazole and substituted triazole derivatives.31, 32 During von Pechmann’s investigations of “osotetrazones” - derivatives of osazones - he was able to determine the actual structure of a 1,2,3-triazole. He did this by heating a compound derived from dimethylglyoxal (C16H16N4) with nitric acid to obtain a colorless oil (C10H11N8). Another reaction using dimethylphenylosazone, derived from phenylhydrazine and a sugar, upon long heating with acid produced C6H5NH2 as the side product. From these studies, von Pechmann proposed that the products formed contained a common unsaturated ring, C2H2N3, as shown in Figure 4. The compounds that von Pechmann investigated were called “osotriazoles”, a designation which is still retained for certain 2-substituted triazole derivatives.33 Examples of reactions formulated by von Pechman are shown below in Scheme 5. H N N N HC CH V Figure 4. von Pechmann's proposed unsaturated triazole ring. 12 H N N N N H H3C (CH3CO)2O C N C C C CH3 H3C CH3 H3C CH3 C N C C (CH3CO)2O N + N 4,5-dimethyl-2-phenyl-2H-1,2,3-triazole Biacetyl di(phenylhydrazone) HO NH2 N N N C N H H3C aniline + H2O N CH3 (2E,3E)-3-(2-phenylhydrazono)butan-2-one oxime 4,5-dimethyl-2-phenyl-2H-1,2,3-triazole Scheme 5. Example reactions by which von Pechamnn made 1,2,3-triazoles.33 Von Pechmann was very perceptive and realized that the compounds he was isolating were actually derivatives of a compound having the formula C2H3N3. Once he realized this, he set out to prepare the parent compound via a degradation reaction.33 Starting with 2-phenyl-1,2,3triazole-4-carboxylic acid (obtained by the oxidation of the 4-methyl derivative31) he first oxidized this compound with potassium permanganate, converting the methyl group on the triazole to a carboxylic acid. This intermediate was next nitrated in the para position of the phenyl group. The 1,2,3-triazole-4-carboxylic acid was obtained by first reducing the nitro group to an amino group by utilizing a mixture of tin (II) chloride and hydrochloric acid. This aminated phenyl compound was removed by oxidative cleavage followed by a decarboxylation which led to the desired unsubstituted triazole (Scheme 6). 13 HC C H3C N alkaline KMnO4 N N HC N C N C HO HNO3 HC N HO O H C HEAT N - CO2 HC N C H C alkaline KMnO4 NH C N O N SnCl2 HCl HC HO NO2 N N O HO NH C N C C N NH2 N N O Scheme 6. Von Pechmann synthesis of 1H-1,2,3-triazole.31, 33 J. A. Bladin34 later showed that the same ring system was present in the benzotriazole compounds. He managed to remove the benzene ring of 5-methylbenzotriazole by oxidation, resulting in a 1,2,3-triaozle-carboxylic acid. This compound was then decarboxylated to give the unsubstituted triazole (Scheme 7), identical to the one obtained by von Pechmann. H N H N N N H3C N H N N HEAT at 200oC alkaline KMnO4 C O C C N HC C N CH O OH HO Scheme 7. Bladin synthesis of 1H-1,2,3-triazole.34 After von Pechmann’s work, a large number of publications covering the 1,2,3-triazole appeared. These reactions ranged in complexity in how the 1,2,3-triazole was obtained. Much of 14 the work involved extensions of Hofmann’s diazotization synthesis of fused-ring 1,2,3-triazoles and von Pechmann’s osazone synthesis. Among the important investigations were those of Dimroth and Fester,35 who discovered that the combination of hydrogen azide or phenyl azide reacting with acetylene formed 1H-1,2,3-triazole and 1-phenyl-1,2,3-triazole, respectively, resulting in the first Click Chemistry reactions. The Dimroth and Fester35 method required acetylene to be dissolved in acetone, hydrogen azide, and absolute alcohol. The mixture would react in a sealed tube at 100oC for 70 hours (Scheme 8). Later, it was found that the combination of phenyl azide with acetylene under similar conditions proceeded more easily, requiring only 40 hours of heating time. H N N HC CH acetylene + HN3 hydrogen azide CH N CH 1H-1,2,3-triazole Scheme 8. Acetylene and hydrogen azide producing 1H-1,2,3-traizole.35 Other methods used to obtain the 1H-1,2,3-triazole required the use of compounds already containing a triazole ring. This was accomplished by reduction,36, 37 oxidation,37 or heating33, 38-40 a substituted triazole to form the 1H-1,2,3-traizole. More elaborate methods were designed to synthesize a 1H-substituted-1,2,3-triazole as well as the 1,4- and 1,5- disubstituted triazoles.41 These methods utilized various alkenes, alkynes, azides, salts and high-pressured reactions. Many of these reactions were accomplished through the use of metal catalysts 39, 42 such 15 as palladium and manganese. Synthesizing regioselective triazoles was also accomplished by using an alkali salt,41 by employing the Grignard43 reaction, or by utilizing alkoxides44-47 to selectively produce either 1,4- or 1,5- disubstituted triazole.37 Since Dimroth and Fester35 in 1910, highly considered methods were devised to synthesize various 1,2,3-triazoles. Many of the reactions employed utilized harsh conditions and carcinogenic solvents. The approach to synthesizing 1,2,3-triazoles using an alkyne and azide was not frequently used until the late 1950’s. The reaction of acetylene and hydrogen azide performed by Dimroth and Fester is an early example of a 1,3-diploar cycloaddition reaction (Scheme 8). The concept of the 1,3-dipolar cylcoaddition, often referred to as a “Huisgen Cycloaddition” reaction, was truly developed and expended by Rolf Huisgen. This method popularized the idea of creating five-membered heterocyclic rings in a more direct route. After his publication in 1955,23 research in Huisgen group immediately boomed, leading to two review articles published in 1963.48, 49 These articles illustrated various reactions using the 1,3-dipolar cycloaddition principle. Since then, research in the synthesis of 1,2,3-triazole derivatives has increased exponentially. The ease of synthesizing a triazole made it easier to research possible applications for the 1,2,3-triaozle derivatives. The disadvantage to this approach was that using an unsymmetric alkyne would lead to different regioisomeric products (Scheme 9). Since there was not a universal method that would guarantee regioselectivity from these reactions, the problem of how to favor the formation of the 1,4- versus the 1,5-disubstituted products became a focus of much research. 16 1 N O + N N N N N 5 4 + O 1 N N 5 O N 4 1,4-isomer 1,5-isomer Scheme 9. Example of regioisomeric products when using asymmetric alkyne. In 2002, Meldal and co-workers found a way to synthesize 1,4-disubstituted-1,2,3triazoles selectively using a copper-(I)-catalyzed reaction.50 This method was typically done with an alkyl azide and an alkyne under either ambient or heated conditions. Through this approach, the synthesis of a regioselective triazole could be accomplished in high yield. Even though these reactions could be performed using copper (I) such as copper iodide or copper bromide, 50, 51 Meldal found that using a mixture of copper (II) (i.e. copper(II)sulfate) and a reducing agent (i.e. sodium ascorbate) to produce copper (I) in situ worked infinitely better.50 Although this method produced a triazole from an alkyne and an azide, it was no longer formally a 1,3-dipolar cycloaddition reaction. This type of reaction was better classified as a Copper-(I) catalyzed Azide-Alkyne Cycloaddition (CuAAC),52 since the copper (I) readily coordinates with the alkyne in the presence of a mild base (i.e. K2CO3) in aqueous solution (Scheme 10), in an approach which resembles a Sonogashira reaction.50, 53 17 N R2 N N R' R1 C C H CuLx H ' R CuLx H+ 5 N N N H H+ 1 R2 R' R1 C C C C CuLx CuLx R2 N 4 2 N C R1 R2 R2 N N N N N CuLx 3 R' C N N C CuLx Scheme 10. Copper-(I) catalyzed Azide-Alkyne Cycloaddition (CuAAC) mechanism. The blue in denoted for the alkyl azide and the red is denoted for the alkyne.50, 53 The very success in the selective formation of the 1,4-isomer using CuAAC highlights the need for selective access to the complementary regioisomer, the 1,5-disubstituted-1,2,3triazole. Although 1,5-disubstituted triazoles were successfully formed by Kleinfeller in 1931,43 as well as L’Abbe in 1969,56 using bromomagnesium acetylides and organic azides, it lacked the scope and the convenience of the CuAAC process.52 The very successful CuAAC was excellent for synthesizing the 1,4-regioisomer, but did not product the complementary 1,5-disubstituted triazole. To achieve this, Sharpless and coworkers utilized several different ruthenium complexes 18 to produce either the 1,4- or the 1,5- disubstituted triazole selectively in “perfect” 100% yield, suggesting complete regiochemical control (Scheme 11).52 N3 Ru + Ru(OAc)2(PPh3)2 CpRuCl(PPh3)2 Cp*RuCl|PPh3)2 Cp*RuCl|NBD) N N N N N N + 1a 1b 85% 100% 100% 100% 15% ------- Scheme 11. Ruthenium-Catalyzed Cycloaddition of Benzyl Azide to Phenylacetylene. 52 There have been many creative methods devised to synthesize 1,2,3-triazoles ranging from complex, step-by-step processes of structural transformation, to the simple cycloaddition of an azide and an alkyne. Since von Pechmann’s discovery of them in 1888, there has been a rapid growth and interest in synthesizing triazoles which stems from the desire to create compounds for use in antifungal drugs, plant protection, anti-allergenics, obesity treatment and osteoarthritis drugs. The catalytic processes discussed seem to offer an unprecedented level of selectivity, reliability, and scope for these synthetic endeavors. However, the success observed using a metal catalyst reduces the “greenness” of these reactions. Prolonged exposure to these metallic salts (either copper or ruthenium) can cause memory loss, increased allergic reactions, high blood pressure, depression, irritability, poor concentration, aggressive behavior, sleep disabilities, fatigue, speech disorders, high blood pressure, autoimmune diseases, and chronic fatigue are just 19 some of the many conditions resulting from exposure to toxins.57 In addition, these reactions often employ organic solvents and metallic salts that are highly toxic and harmful to the aquatic environment. With these concerns came a growing interest in reducing or eliminating the use and generation of substances hazardous to human health and the environment. The field of Green Chemistry was created to encourage the design, development, and implementation of chemical products and processes to reduce or eliminate the use and generation of hazardous substances. Improving research and development productivity has been one of the biggest problems in designing more environmentally friendly processes. The bottleneck for conventional combinatorial synthesis was the optimization of reaction conditions to afford the desired products in suitable yields and purities. Since many reaction sequences require at least one or more heating steps for extended time periods, these optimizations are often difficult and time-consuming. Microwave-assisted heating has been shown to reduce the need for solvents and catalysts, making the reaction “greener” and more environmentally friendly. 20 Brief History for the Discovery and Optimization of Microwave Chemistry The development of the microwave was stimulated by World War II when a magnetron was designed to generate a fixed frequency of microwaves for Radio Detection and Ranging (RADAR) devices.58, 59 In 1946, an engineer by the name of Dr. Percy Spencer came across something interesting when studying the magnetron. He noticed that the microwave energy was able to cook food when a candy bar in his pocket melted during one of his magnetron experiments. Further studies showed that microwaves could increase the internal temperature of foods much more quickly than a conventional oven.60 This drove companies to invest in the idea of a commercial microwave for household use in 1954. Investigation into industrial applications for microwave energy also began in the 1950’s, and has led to applications including the removal of sulfur and other pollutants by the irradiation of coal,60 rubber vulcanization,60 product drying,60, solvent and compound extraction.65, 66 61 moisture and fat analysis,62-64 along with As improvements and simplifications increased, the purchase cost for the microwave decreased, allowing it to become an affordable, common household item. Investigation into modifying and applying the domestic microwave oven for scientific research was not examined until the late 1980’s. In 1988, there were two papers published on microwave-assisted reactions,67, 68 and since then, many chemists have discovered the benefits of using microwave energy to drive synthetic reactions. In 2010, more than 16,000 papers regarding microwave-assisted chemistry were published. Many reactions were performed using a household microwave until problems arose. These household microwave ovens were not designed for rigors of laboratory use since there were no built-in safety controls. Acids and solvents quickly corroded the interior of the microwave, and the ability to regulate temperature and pressure had not yet been developed. In 21 addition, the cavity of the microwave was not designed to withstand explosions that occurred when vessels failed during runaway reactions.60, 69 These problems encouraged companies to manufacture a multi-mode microwave oven that addressed all of these issues. These laboratory grade microwaves worked well for large-scale applications, but had some fundamental limitations in performing small-scale synthesis. Recently, single-mode technology which provides more uniform and concentrated microwave power has become widely available.60 The use of the microwave oven in synthetic chemistry has increased because of its efficiency, the option for solvent-free reaction conditions, reduced reaction times, enhanced yields, and selectivity. Several of these advantages lend themselves to an eco-friendly approach, termed Green Chemistry.70-72 Green Chemistry has twelve basic principles that act as guidelines for what are environmentally friendly reactions. Among the twelve principle of Green Chemistry, the best principles to facilitate the design of sustainable processes for the chemical industry 73 are shown by performing reactions that are solvent-free, metal-free, and that reduce waste. Microwave-assisted synthesis has now been using in many types of reactions, in particular cycloaddition reactions, which are widely used reactions in organic synthesis. 74, 75 Cycloaddition reactions typically require the use of elevated temperatures and long reaction times, however, the shorter reaction times associated with microwave-assisted reactions makes it a very attractive application. The shorter reaction times lower the possibility for the polymerization and the decomposition of reagents and products which often plague the thermal cyclizations.74, 76 Several methods have been devised using the microwave to assist in the synthetic process for cycloaddition reactions. These reactions have been performed under a variety of conditions including 1) in sealed vessel under pressure,67, 68 2) with refluxing,77, 78 3) 22 under neat or solvent-free,71, 72, 79 4) using mineral supports,80-82 5) without supports,83-85 and 6) heat captors to assist in rapid heating of a reaction.86 From the examples listed above, the versatility microwave-driven syntheses can be easily seen. Microwave-assisted reactions often simplify the process needed to synthesize compounds compared to conventional methods. The rapid heating of the microwave avoids the excessive heating associated with classic heating. In fact, the microwave-assisted approach facilitates 1,3dipolar cycloadditions that are typically difficult, and sometimes impossible, to achieve with the classical approach.87 Successful 1,3-dipolar cycloaddition reactions have been accomplished with various 1,3-dipoles such as azomethine ylides,87 nitrones,88 azomethine imines and azides89 (see Table 1). Synthesizing 1,2,3-triazoles using 1,3-dipolar cycloaddition by microwave irradiation opens up the possibility for the synthesis of various triazoles that once seemed difficult. Many triazoles that have been synthesized classically have also been successfully accomplished using microwave irradiation, producing excellent results compared to classic synthesis. For example, triazoles have been synthesized to contain sulfur,90 phosphorus,91 boron,92 or a metal93 incorporated in the compound at either the N1, C4 and C5 positions. The interest of using the 1,3-dipolar cycloaddtion in the microwave came about at the turn of the 21st century. The first 1,2,3-triazole was successfully synthesized using a household microwave oven in 2001, a year before Meldal published his work on selective synthesis of 1,4disubstituted-1,2,3-triazoles. Tao and co-workers successfully synthesized several 1,2,3-triazoles without a metal catalyst using a domestic microwave oven.94 The reaction simply required an azide, an α-keto phosphorus ylide and recyclable silica gel (as a support) in an open vessel which was irradiated for 4-10 minutes at 400 Watts with moderate to excellent yields (Scheme 12). 23 Ar N Ar-N3 + R-CO-CH=PPH3 MW , 4-10 min. silica gel C R N N C H 60-90% Scheme 12. Formation of a derivative of 1,2,3-triazole using an azide and an ylide.94 Interestingly enough, the α-keto phosphorus ylides used may be seen as having internal stereocontrol for the formation of triazoles, since all of the triazoles produced were the 1,5disubstituted regioisomer. This was an interesting finding because it illustrated two important synthetic features. The first, formation of 1,2,3-triazole in a microwave oven and the second, regioselective synthesis. This could very well have been the first green triazole synthesis employing a microwave and exhibiting regioselectivity. The significance of the work from both Meldal and Tao shows that microwave-assited heating could reduce heating times of classical cycloaddition reactions from hours to a matter of minutes with similar or often better results. Later, in 2004, Van der Eycken and co-workers were able to synthesize various 1,2,3triazole derivatives using copper (I) catalysis in the microwave oven. 95 This synthetic approach formed only the 1,4-disubstituted triazole isomer and reduced the reaction time tremendously compared to the previous CuAAC approach utilizing conventional methods. The copper (I) catalyzed reaction for this kind of transformation has placed it in a class of its own and has enabled many novel applications.96 Similarly to Meldal’s approach, Van der Eycken had to use a mixture of copper (II) and a reducing agent to produce copper (I) in situ for the reaction to proceed successfully with no trace of the 1,5-regioisomer. In that same year, Van der Eycken97 24 was able to synthesize 1,2,3-triazole derivatives using terminal acetylenes and glycosyl β-azides in the presence of copper (I), which resulted in moderate to high yields of 30-90%. Ruthenium catalysis, which was used to selectively synthesize the 1,5-regioisomer in classical synthesis, has not yet been shown to be successful in microwave reactions. 25 Statement of Problem The primary goal of this research was to synthesize various 1,2,3-triazoles using a domestic microwave oven and without the use of organic solvent, catalysts or extended heating. This will serve to emphasize the significance of green chemistry. The synthetic method chosen provided a fast and efficient technique to obtain the desired triazoles and afforded a good alternative to the classical synthesis without the use of toxic and environmentally unsafe reagents. 26 Chapter 2 RESULTS AND DISCUSSION – BACKGROUND ANALYSIS The purpose of this work was to design a synthetic method that could be used to synthesize a wide variety of simple and complex 1,2,3-triazoles under the greenest conditions possible. Synthesis of these triazoles was done by microwave heating a mixture of an alkyne and an azide utilizing minimal to no solvent (Scheme 13). In designing methods for the formation of 1,2,3-triazoles, two main principles have been used as guidelines: 1) preventing waste is better than cleaning it up, and 2) less toxic alternatives should be used wherever possible. To adhere to these principles, the study was focused on reactions that can be run in the absence of solvents (thereby eliminating the major source of waste in any reaction) and without added catalyst, since many catalysts are toxic, metal-based compounds. These reactions were also designed to be performed in an inexpensive, domestic-style microwave oven to avoid the high costs of the laboratory-grade microwave. R R N3 + 2 R R3 mw N N R2 N R3 Scheme 13. 1,3-Dipolar cyclization forming a 1,2,3-triazole, where R, R2 and R3 are carbon based substitutents. 27 Typically, triazole-forming cycloaddition reactions have been attempted under a wide variety of conditions, with varying degrees of success. Triazoles have be synthesized using thermally driven cyclizations. However these reactions are generally slow and require high temperatures, often utilizing steel bomb reactors.98-101 Triazole formation has also been successfully accomplished using metal-based catalysis in reactions which exhibit both high yields and regioselectivity.50, 52, 95 The disadvantage to this approach is that these reactions often employ organic solvent, such as dimethyl sulfoxide (DMSO), and that the metallic salts utilized as catalysts are highly toxic and harmful to the aquatic environment. Therefore a “greener”, more eco-friendly, procedure would be desirable. The utilization of microwave-assisted synthesis reactions offers several advantages over standard heating methods. Not only are microwaveassisted reactions faster than their classical counterparts, they are generally cleaner, require less energy to run, and allow for easy heating of small-scale reaction mixtures. The use of standard laboratory glassware and a domestic microwave oven keeps the costs associated with these experiments much lower than procedures that utilize laboratory grade microwave ovens and specialty glassware. The reactions performed in this study demonstrate that various 1,2,3-triazoles could be generated without the use of conventional heating or the use of expensive mono-mode microwave ovens. The reactions in this study were performed primarily in a domestic-model microwave oven using standard glassware to show that microwave assisted organic synthesis can be accomplished without specialized materials, illustrating that this method was capable of producing several 1,2,3-triazoles with equal or better results compared to those in literature. In this study, benzyl azide was used extensively to create several 1,2,3-triazoles. However, it was also of interest to observe if this method may be applied to other azides, rather than just benzyl azide. The goal of using different azides would be to illustrate the diversity and versatility of this developed 28 technique. The production of different azides was accomplished by using known and accepted literature methods, these azides were ethyl 2-azidoacetate, 2-(2-Azidoethoxy)ethanol and 1,4bis(azidomethyl)benzene (Figure 5). Some 1,2,3-triazoles synthesized in this study were not found in the literature, and required more advanced spectral analysis (i.e. Nuclear Overhauser Effect, nOe) to determine the regioisomer isolated. The goal of the study presented here was to illustrate how the synthesis of pharmaceutically useful triazole moieties could be accomplished easily and in moderate to high yields without use of expensive laboratory equipment. O N3 N3 O benzyl azide N3 ethyl 2-azidoacetate O HO N3 2-(2-Azidoethoxy)ethanol N3 1,4-bis(azidomethyl)benzene Figure 5. Various azides used in this study. Initial microwave explorations were performed by setting the microwave at 30% power and heating for 3 minutes. The starting materials were all placed in an Erlenmeyer flask and loosely covered (Figure 6). This design was incorporated for all reactions to allow for some venting, because heat and pressure can build up during the reaction. This design allowed for the excess heat and pressure to be released safely. 29 Figure 6. Glassware used for the optimized reactions. Optimization of each reaction was conducted to determine the proper power setting and reaction time needed in order to achieve optimal results. Variations in the power settings and times for each reaction may have been due to the fact that the power levels on a domestic microwave oven are not controlled and do not indicate an actual change in power output. Rather, a setting of 30% power indicates that the microwave will be on at full power but only irradiating the sample 30% of the time. The remaining 70% of the time, the sample is rotating on the carousel absorbing the remaining microwaves that may be reflecting inside the microwave cavity. Thus, microwaves of wattages under identical power level settings will have significant differences in power output. It was also noted that the amount of power which needed to be applied to the mixture varied with the subsituents on the alkyne. From classical synthetic methods, it was observed that the ease of the azide cyclization is governed by the polarization of the acetylene. It was found that when the electron density of the alkyne was reduced, (i.e. when the alkyne carried strong electron withdrawing groups), the reaction reached completion faster with the least amount of power 30 output from the microwave. The opposite was true when the alkyne had substituents that were electron donating – rich alkyne required the most power output and took longer to reach completion. Some of the results obtained for the single ring systems were the preliminary reactions explored during undergraduate work. The results showed a variation of yields that were obtained depending on the chosen starting materials. This preliminary data provided the background needed for the graduate study. Preliminary studies illustrated that a 1,3-dipolar cycloadition reaction with a domestic microwave oven. The data obtained here were necessary to provide comparisons for the reactivity differences between the single and double ring systems. 31 Single 1,2,3-Triazoles 1,3-Dipolar cycloaddition reactions were originally believed to work only if the alkyne contained two electron withdrawing substituents. However, this theory was quickly proven to be incorrect as preliminary studies showed that any alkyne could undergo this cyclization in a domestic microwave to some degree. The biggest disadvantage to working with alkynes that were more electron rich was the increased reaction times and applied power necessary to drive the reactions. This discovery broadened the scope of the possible triazoles that may be synthesized with relative ease. The collected results from this study of 1,3-dipolar cycloaddition reactions of a variety of alkynes and with azides are shown in Table 2. Table 2. 1,3-Dipolar cycloaddition reactions between an alkyne and azide to produce simple 1,2,3-triazoles. Entry Azide Alkyne Reaction Conditions Products Yield CH2Ph 1 N3CH2Ph H C C CO2H N N 30% power/5 min 99% H N H PhH2C N 2 N3CH2Ph Ph C C Ph 80% power/15 min Ph 80% N N Ph CH2Ph H3CO2C 3 N 30% power/30 sec N3CH2Ph H3CO2C C N C CO2CH3 98% N H3CO2C CH2CO2CH2CH3 H3CO2C 4 N3CH2CO2CH2CH3 H3CO2C C C CO2CH3 30% power/10 sec N H3CO2C N H3CO2C 5 TMSN3 H3CO2C C C CO2CH3 87% N H N 30% power/6 min N H3CO2C 61% N 32 33 Using symmetric alkynes (i.e. alkynes containing the same functional groups on each end of the molecule) made it easy to determine the success of the reaction because there were no concerns about isomer formation. An interesting result of early inestigations suggested that microwave heating could not only drive the desired cyclization reaction, but could also provide unexpected results. The reaction between benzyl azide and propiolic aicd (entry 1, Table 2) was expected to result in the acid-substitution triazole. Instead it produced unsubstituted 1-benzyl1,2,3-triazole. The change in functional group from the carboxylic acid would result from a decarboxylation, which is commonly thermally driven. The loss of carbon dioxide from βcarboxylic acid shown in Scheme 14. O H3C H O O heat OH H3C O OH O O + H3C β-keto acid tautomerization CH2 enol O C O H3C CH3 keto Scheme 14. Decarboxylation of a β-carboxylic acid. Typically, the decarboxylation of simple carboxylic acids are considered difficult and are rarely encountered.102 However, it was of interest to see that under this greener approach to 1,3dipolar cyclization which is done without solvents, buffering solutions or protecting groups, an unexpected decarboxylation did occur. Reported triazole cyclizations using carboxylated alkynes show no evidence of a decarboxylation reaction accompanying the triazole formation.1 It is believed that this decarboxylation is thermally driven, as the method employed did not include 34 any attempt to control the heat of the cyclization reaction. The 1H NMR spectrum of this decarboxylated product is shown in Figure 7 below. SpinWorks 2.3: B Ph A H 2C N N H N B C A H C PPM D D 7.6 7.4 7.2 7.0 file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 3\2005\120905_CR98\1\fid expt: <zg30> transmitter freq.: 300.144690 MHz time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 16 6.8 6.6 6.4 6.2 6.0 5.8 5.6 5.4 5.2 freq. of 0 ppm: 300.142604 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 Figure 7. The 1H NMR of 1-benzyl-1,2,3-triazole. From the 1H NMR, it was observed that the carboxylic acid was not present in the expected downfield range of about 11.4 ppm. The -CH2- peak from benzyl azide was shifted from 4.4 ppm in the starting material to 5.6 ppm in the product. Chemical shifts for this product were compared to literature spectral data and results were comparable.12 The decarboxylation was believed to take place after the triazole formation, since if propiolic acid decarboxylated prior to cyclization it would produce acetylene, boiling point -83.3oC, which would have vaporized before it could react with benzyl azide since no effort was taken to seal the reaction vessel. Thus, it is believed that the carboxylic acid acted as an auxiliary group, activating the alkyne to react quickly under these simple conditions, before detaching to leave the unactivated triazole product. 35 In this way, it could be possible to produce triazoles which appear to originate from electron-rich terminal alkynes. Another interesting point to highlight was the result from the use of electron-rich alkynes in this 1,3-dipolar cyclization reaction. Diphenylacetylene has been reported to be sluggish or unreactive towards triazole formation in the absence of an outside catalyst.52 However, using this method, diphenylacetylene reacted smoothly with benzyl azide to provide 4,5-diphenyltriazole in high yields under simple heating by a domestic microwave oven, giving comparable results to literature data (entry 2, Table 2). This reaction required a microwave heating power of 80% and irradiation of the mixture of benzyl azide and diphenyl acetylene for 15 minutes in order to obtain the desired product (Figure 8). Typically, reactions utilizing more electron-rich alkynes required more intense heating, causing a greater loss of compounds (possibly due to evaporation). This lead to slightly lower yields for these products as the heating times become longer. SpinWorks 2.3: B Ph B, C & D A CH2 N N N Ph C Ph D PPM 7.8 7.6 7.4 7.2 7.0 6.8 file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 3\2006\042806_CR146\1\fid expt: <zg30> transmitter freq.: 300.144690 MHz time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 16 6.6 6.4 6.2 6.0 5.8 A 5.6 freq. of 0 ppm: 300.142607 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 Figure 8. The 1H NMR of 1-benzyl-4,5-diphenyl-1,2,3-triazole in CDCl3. 5.4 5.2 5.0 4.8 36 Figure 8 shows the 1H NMR of 1-benzyl-4,5-diphenyl-1,2,3-triazole produced by this reaction. The product was analyzed by 1H NMR immediately after the reaction completed without further purification. The -CH2- peak (A) for the starting material was no longer present at 4.31 ppm and had shifted to 5.40 ppm. The aromatic region for this triazole was integrated and was found to integrate to 15 protons as expected. D D D D SpinWorks 2.3: D D E B-E E E x 1.000 144.0 140.0 136.0 132.0 C C N E E N N A B B B B E B B CDCl3 128.0 A PPM 135.0 125.0 115.0 file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 3\2006\042806_CR147\1\fid expt: <zgpg30> transmitter freq.: 75.478523 MHz time domain size: 65536 points width: 17985.61 Hz = 238.287803 ppm = 0.274439 Hz/pt number of scans: 1024 105.0 95.0 85.0 75.0 65.0 55.0 freq. of 0 ppm: 75.470919 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 Figure 9. The 13C NMR of 1-benzyl-4,5-diphenyl-1,2,3-triazole in CDCl3. Based on the 13 C NMR, it was observed that the alkyne carbons were absent from the spectrum, as shown in Figure 9. If alkyne carbons were still in the crude mixture, they would be found at around 90 ppm. In addition, the -CH2- carbon of the benzyl azide starting material would typically be found at 60 ppm. The -CH2- in the product triazole was observed upfield from this 37 position, around 52 ppm. The chemical shifts of the aromatic carbons were compared to literature and were found to agreed well.52 Thus, through these findings, it was concluded that 1-benzyl4,5-diphenyl-1,2,3-triazole was successfully synthesized in 80% yield. Also included in the preliminary studies was an exploration of the affects of azide substitutions on the cyclization reaction. In these reactions, azides that were either electron withdrawing or donating were used to study the effects on the reaction to the production of triazoles. Several attempts were made to create new azides that could be used to synthesized novel triazoles, since the reactions performed utilizing benzyl azide (an electron donating 1,3dipole) had worked so successfully. Only a few of these azides produced triazole products in good yields. The two azides that were successful in producting triazoles under neat conditions were ethyl 2-azidoacetate and trimethylsilyl azide. In the reactions with dimethyl acetylenedicarboxylate, all of the azides tested gave moderate to high yields with their corresponding triazoles (61-98%). The reaction of benzyl azide with dimethyl acetylenedicarboxylate (entry 3, Table 2) was successful, producing virtually quantitative yields of the desired product. Analysis of the crude product by 1H NMR in CDCl3 and showed the methyl esters (C & D) as two peaks, each integrating to 3H (Figure 10). When the product was analyzed in d6-DMSO, an unexpected outcome arose. In d6-DMSO, both of the methyl ester signals appeared as one peak at about 3.84 ppm, suggesting a triazole that was symmetric (Figures 11). 38 SpinWorks 2.3: B A Ph H2C C H3CO2C N N N H3CO2C D C D 4.0 3.8 A B PPM 7.4 7.2 7.0 6.8 6.6 6.4 6.2 6.0 5.8 file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 1\040805_CR170\1\fid expt: <zg30> transmitter freq.: 300.144690 MHz time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 16 5.6 5.4 5.2 5.0 4.8 4.6 4.4 4.2 freq. of 0 ppm: 300.142603 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 Figure 10. The 1H NMR of dimethyl-1-benzyl-1,2,3-triazole-4,5-carboxylate in CDCl3. SpinWorks 2.3: B A Ph H2C C H3CO2C N N D H3CO2C C, D A B PPM N 7.2 7.0 6.8 6.6 6.4 6.2 file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 1\040905_CR173\1\fid expt: <zg30> transmitter freq.: 300.144690 MHz time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 16 6.0 5.8 5.6 5.4 5.2 5.0 4.8 4.6 4.4 4.2 4.0 freq. of 0 ppm: 300.142597 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 Figure 11. The 1H NMR of dimethyl-1-benzyl-1,2,3-triazole-4,5-carboxylate in d6-DMSO. 3.8 39 It is believed that the overlapping signals in d6-DMSO are fortuitous (Figure 11), and do not reflect the actual structure of the molecule. It is not likely the benzyl group would be able to shift from the 1-N nitrogen to the 2-N nitrogen position (as a proton would be capable of doing), since a benzyl group is much larger and is held by a stronger bond. It is more likely that the benzyl group resides on the 1-N position. The -CH2- group (A) was found to be a singlet since there were no neighboring hydrogens, and in both solvent systems the -CH2- was found around 5.80 ppm. The reaction of dimethyl acetylenedicarboxylate with ethyl 2-azidoacetate (entry 4, Table 2) was also attempted with great success. The crude product was analyzed by 1H NMR and the spectrum showed that product was very clean without any need for further purification. The 1 H NMR spectrum for this product is shown in Figure 12 below. SpinWorks 2.3: O B A C O O N N O E N E O D O D C A B PPM 5.2 4.8 4.4 TMS 4.0 3.6 file: C:\Documents and Settings\Cat Roush\My Documents\Grad Class\Chem 294\Triazole Spectra\BOOK 3\2006\061506_cr125\1\fid expt: <zg30> transmitter freq.: 300.144690 MHz time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 16 3.2 2.8 2.4 2.0 1.6 1.2 0.8 0.4 -0.0 freq. of 0 ppm: 300.142597 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 Figure 12. The 1H NMR of dimethyl-1-(2-ethoxy-2-oxoethyl)-1,2,3-triazole-4,5-carboxylate in CDCl3. 40 From Figure 12, the 1H NMR spectrum looks rather simple, but tells a lot about the product synthesized. The CH3 (A) and CH2 (B) protons from the ethyl group were found at 1.30 ppm and 4.26, respectively. Since these protons did not shift much from their location in the starting material, then NMR did not immediately suggest that the product was formed. The methyl ester peaks, D and E, were found as two peaks each integrating to 3H and found 3.99 ppm, not far from the original 3.85 ppm in the starting material. The biggest change upon formation of the product triazole occurred with the -CH2- peak (C). The starting material, ethyl 2azidoacetate, showed a -CH2- peak at 3.97 ppm, while the product 1H NMR shows this peak shifted further downfield to 5.45 ppm. Through these findings, it was concluded that dimethyl-1(2-ethoxy-2-oxoethyl)-1,2,3-triazole-4,5-carboxylate was successfully synthesized in 87% yield. Another interesting reaction was observed in the reaction of trimethylsilane azide (TMSN3) and dimethyl acetylenedicarboxylate. In the reaction of TMS-N3 with dimethyl acetylenedicarboxylate, the analysis of the crude product mixture immediately after heating indicated that no TMS group was present. The 1-H triazole (entry 5, Table 2) was successfully isolated in 61% yield. Typically, the removal of a trimethyl silyl (TMS) group would be accomplished by nucleophilic displacement with fluoride or oxygen nucleophiles (i.e. water), however neither of these should have been present in the reaction system. To confirm the initial analysis, the crude product was dissolved in diethyl ether and washed with sodium bicarbonate to help purify the product further. The 1H NMR spectrum from the initial analysis and the extracted product were the The 1H NMR showed the N-H (A) as a very broad peak at 16.28 ppm, as shown in Figure 13. 41 A A B H3CO2C H N x 256.000 16.4 16.0 B 15.6 B N H3CO2C N TMS PPM 16.0 15.0 14.0 13.0 12.0 11.0 10.0 9.0 8.0 7.0 6.0 5.0 4.0 3.0 2.0 1.0 0.0 -1.0 file: C:\DocumentsandSettings\Cat Roush\My Documents\Under GradClass\CHEM189&198\Paper PublicationData\Dr KNMRspectraanddata\1H_diester inDMSO\1\fid expt: freq. <zg30> of 0ppm: 300.142603MHz transmitter freq.: 300.144690MHz processedsize: 32768complexpoints timedomainsize: 65536points LB: 0.000 GB: 0.0000 width: 6172.84Hz=20.566213ppm=0.094190Hz/pt number of scans: 16 Figure 13. The 1H NMR of dimethyl-1H-1,2,3-triazole-4,5-carboxylate, with an insert showing he N-H peak region from 15.6 – 16.6 ppm. The observed peaks for the -CH3- groups (B) of the two methyl esters were seen at 3.95 ppm (Figure 13). The observed peak for the N-H was a broad peak due to the averaging signals of the two triazole tautomers. This broad peak illustrated that the rate of proton exchange from N1 to N2 is extremely fast (Figure 14). The δ-values for the methyl esters in 1-H-1,2,3-triaozle4,5-dicarboxylate were identical (B) because of the fast equilibrium between the 1-H and 2-H tautomers.7 H N N 1 O O O N O O 1-H tautomer N H N2 N O O O 2-H tautomer Figure 14. Dimethyl-1H-1,2,3-triazole-4,5-carboxylate. 42 Had the TMS group remained intact and localized on the 1-H position, the 1H NMR analysis would have shown a single sharp peak integrating to nine protons. These methyl groups would have been quite shielded and would have been observed near 0 ppm. Since this was not observed, it was postulated that the loss of the TMS group occurred during an intermediate step in the process of forming the 1,2,3-triazole. The reaction was believed to proceed via a 1,3-dipolar cyclization as shown in Scheme 15. H2 C CO2CH3 C Si(CH3)3 (H3C)2Si H N C CO2CH3 H N N N C CO2CH3 N N N N CO2CH3 Si + N C CH2 CO2CH3 H3C CH3 CO2CH3 dimethylmethylenesilane Intermediate Step Scheme 15. Purposed mechanistic pathway affording dimethyl-1H-1,2,3-triazole-4,5-carboxylate. Typically, TMS deprotection reactions require the presence of a nucleophile to drive the TMS group off of the nitrogen. However, the reaction condition employed was neat and did not include an available nucleophile source. Thus, the suggested mechanism, shown in Scheme 15, proposes that a proton from one of the nearby methyl groups on the silicon atom was donated to the nitrogen during an elimination reaction. The resulting side product would then be dimethylmethylene silane, a low-boiling point compound which would have vaporized during heating. This would explain why none of it was recovered or observed in the 1H NMR of the crude or purified product. 43 Chapter 3 RESULTS AND DISCUSSION – CURRENT WORK Isomers of Simple 1,2,3-Triazoles Preliminary studies into the formation of substituted 1,2,3-triazoles using a domestic microwave oven showed that the approach could provide products in high yields and with some interesting and unexpected results. These results provided a basis for the present study which focused on several new aspects to this reaction method. These new aspects include the possibility of regioisomer formation and the ability to form multiple triazole rings from a difuntionalized substrate. It was of interest to synthesize more complex 1,2,3-triazoles to observe the versatility of this method and compare results for reactions that have been successfully completed and published in the literature. When using an unsymmetrically substituted alkyne, there are actually 2 possible regioisomers which can be produced in the dipolar 1,3-addition reaction (Scheme 16). The method employed in this study does not attempt to control the regiospecificity of the sysnthesis of 1,2,3-triazoles. Many 1,2,3-triazoles published today, which afford regioselectivity, have utilized metal-based complexes to synthesize a single desired isomer.50, 96 However, if metal-based complexes are not used and no attempt to control the reaction is undertaken, the result should be a mix of both 4- and 5-substituted isomers. It is important to note that even though the product should theoretically produce a 1:1 ratio of each isomer, there is always the possibility that one isomer will predominate due to steric hinderence. The overall yields of the several nonregiospecific reactions for the synthesis of substituted 1,2,3-triazoles are summarized in Table 3. 44 1 N O + N N N N N 5 + 4 O 1,4-isomer Scheme 16. Example of regioisomeric products when using asymmetric alkyne. 1 N 5 N N O 4 1,5-isomer Table 3. 1,3-Dipolar cycloaddition reactions between an alkyne and azide to produce non-regiospecific 1,2,3-triazoles. Entry Azide Alkyne Reaction Conditions Products CH2Ph H 1 N3CH2Ph H C C Ph N N N H 24.2 % + 2 N3CH2Ph HO2C C C Ph N N N H 32 % + 3 N3CH2Ph Ph C C CO2CH2CH3 N 54.1% 1 : 2.9 68.7% 1.1 : 1 N N + N H3CH2CO2C 1 : 2.3 CH2Ph H3CH2CO2C N 30% power/6 min 46% 14 % CH2Ph Ph 2.5 : 1 N + N Ph 46% CH2Ph Ph N 30% power/6 min 1:1 24.1 % CH2Ph H 48.3% N + N Ph Ratio CH2Ph Ph N 80% power/9 min Yield N Ph 14 % + 32 % CH2Ph H CH2Ph N N 4 N3CH2Ph C C H + N N N + H H C C CH2OH N 30% power/6 min N HOH2C + O HOH2C N H N N N 36.1 % 40.1 % OH O N3CH2CH2OCH2CH2OH N H 14.0 % 5 N N N 50% power/9 min + OH 32.6 % 45 46 One of the first molecules to be studied arose from the reactions of phenylacetylene and phenylpropiolic acid with benzyl azide (entries 1 & 2, Table 3). An interesting observation occurred from the analysis of the products from these reactions. Even though these reactions involve the use of two different alkyne starting materials, both reactions resulted in the formation of the same product – a mixture of 1-benzyl-4-phenyl-1,2,3-triazole and 1-benzyl-5-phenyl-1,2,3triazole. The reaction of phenylproiolic acid and benzyl azide was accompanied by complete decarboxylation in the resulting triazole products. The 1H NMR spectra are shown in Figures 15 and 16. SpinWorks 2.3: C Ph A H B CH2 N N C C Ph N B A C PPM 7.6 7.4 7.2 7.0 file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 3\2006\071106_CR190A\1\fid expt: <zg30> transmitter freq.: 300.144690 MHz time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 128 6.8 6.6 6.4 6.2 freq. of 0 ppm: 300.142605 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 Figure 15. The 1H NMR of 1-benzyl-4-phenyl-1,2,3-triazole in CDCl3. 6.0 5.8 5.6 47 SpinWorks 2.3: C Ph B H2C C Ph N C N N H A B A PPM 7.6 7.4 7.2 7.0 file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 3\2006\071106_CR190B\1\fid expt: <zg30> transmitter freq.: 300.144690 MHz time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 128 6.8 6.6 6.4 6.2 6.0 5.8 5.6 freq. of 0 ppm: 300.142603 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 Figure 16. The 1H NMR of 1-benzyl-5-phenyl-1,2,3-triazole in CDCl3. Previous studies had shown that propiolic acid reacted under these conditions to form a decarboxylated triazole product, the reaction of phenylpropiolic acid was observed for any similarities to the previous reactions. Analysis of the crude reaction product showed that the carboxylic acid was no longer present. Additional studies in which the phenylpropiolic acid was heated by itself in the microwave showed that no decarboxylation occurred in the absence of the triazole ring formation. It was therefore concluded that this reaction occured by first undergoing the 1,3-dipolar cyclization reaction, with the decarboxylation occurring afterwards. This was further supported by the observation that both the phenylacetylene (entry 1, Table 3) and phenylpropiolic acid (entry 2, Table 3) each react with benzyl azide to give the same triazoles with the same overall isolated yield. However, the more polar phenylpropiolic acid produced the 4-subsituted product in a 2.5:1 ratio over the 5-substituted product while the phenylacetylene cyclization produced a 1:1 ratio of these same isomers. Thus, the carboxylic acid group must have 48 stayed on long enough to exert a large influence on the regioselectivity of the dipolar cycloaddition reaction before decarboxylation take place. This study was extended to include the reaction of a lower-polarity alkyne – ethyl phenylpropiolate with benzyl azide (entry 3, Table 3). Two factors made this reaction of potential interest – electronegativity and sterics. The ethyl phenylpropiolate contained an ester which is a strong electron withdrawing group. In previous reactions, electron poor alkynes produced the highest yields of triazole products. In addition, the phenyl group on the alkyne was larger and bulkier than the ester group present on the other side of the alkyne. Hence, steric factors should be more pronounced in the reaction, and should influence how much of each isomer would form. With the reaction of ethyl phenylpropiolate and benzyl azide optimized, the isomers were purified and isolated successfully in an overall yield of 46%. Unfortunately, the higher yields expected with the electron-withdrawing group was not observed. This reaction illustrated that electonegativity was clearly not the only factor which determined how well these reactions would proceed. The 1H NMR spectra for both product isomers are shown in Figures 17 and 18. 49 SpinWorks 2.3: SpinW orks 2.3: D E x A H3C 1.000 7.70 7.60 7.50 B H2 C Ph C D H2C O C O N N 7.40 N Ph E CH2Cl2 D E A C B PPM 8.4 8.0 7.6 7.2 6.8 6.4 6.0 file: C :\ Do cumen ts and Settings\C athleen Roush\ My D ocu ments\Resea rc h\BOOK 2\ 0630 05_C R21 2\1\ fid 5.6 5.2 4.8 4.4 TMS 4.0 expt: <zg30 > transmitter freq .: 300. 14469 0 MH z 3.2 2.8 2.4 2.0 1.6 1.2 0.8 0.4 -0.0 -0.4 -0.8 proce ssed size : 32 768 complex points time d omain size: 655 36 p oints LB: 1 0. 000 GB: 0. 0000 Figure 17. The H 6.4 NMR6.0of ethyl 1-benzyl-4-phenyl-1,2,3-triazole-5-carboxylate CDCl 3. 1.2 7.6 7.2 6.8 5.6 5.2 4.8 4.4 4.0 3.6 3.2 2.8 2.4 2.0 1.6 width: 6 172. 84 Hz = 20.5 6621 3 pp m = 0. 0941 90 H z/ pt PPM 3.6 freq. of 0 ppm: 300 .142 609 MHz numb er of scans: 16 file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 2\063005_CR212\1\fid expt: <zg30> transmitter freq.: 300.144690 MHz time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 16 0.8 freq. of 0 ppm: 300.142609 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 D SpinWorks 2.3: E SpinW orks 2.3: C PhD H2C E Ph N N x H3C O C C A H B2 O 2.000 7.4 7.2 7.0 A C E D N acetone B TMS H2 O PPM PPM 7.6 7.2 6.8 7.2 6.8 6.4 6.4 6.0 6.0 5.6 file: C :\ Do cumen ts and Settings\C athleen Roush\ My D ocu ments\Resea rc h\BOOK 2\ 0630 05_C R21 2A\1 \fid transmitter freq .: 300. 14469 0 MH z 5.6 5.2 e xpt: <zg3 0> file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 2\063005_CR212A\1\fid expt: <zg30> time d omain size: 655 36 p oints transmitter freq.: 300.144690 MHz width: 6 172. 84 Hz = 20.5 6621 3 pp m = 0. 0941 90 H z/ pt time domainnumb size:er 65536 points of scans: 16 width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 16 1 5.2 4.8 4.8 4.4 4.4 4.0 3.6 4.0 3.6 3.2 2.8 3.2 2.4 2.8 2.0 2.4 1.6 1.2 2.0 0.8 1.6 0.4 1.2 freq. of 0 ppm: 300 .142 603 MHz proce ssed size : 32 768 complex points LB: freq. of 0 ppm: 300.142603 MHz 0. 000 GB: 0. 0000 processed size: 32768 complex points LB: 0.000 GB: 0.0000 Figure 18. The H NMR of ethyl 1-benzyl-5-phenyl-1,2,3-triazole-4-carboxylate CDCl3. -0.0 0.8 -0.4 0.4 0.4 -0.0 50 The assignment of each isomer was made by comparison to the findings of Cwiklicki and Rehse.103 Cwiklicki and Rehse reported that the compound which eluted first from the column was the 5-carboxylate, and it was followed by the 4-carboxylate. According to this literature source, the isomer assignments were made from the 1H NMR of the benzylic -CH2- group in the 1-position of the triazole. The paper suggested that if the ester was vicinal to the benzylic group, (i.e. in the 5-position) then there would be an anisotropic effect, causing a downfield shift of the CH2- group to about 5.94 ppm. Having the phenyl group in the vicinal position, (i.e. the ester in the 4-position) resulted in a more upfield position for this methylene signal at about 5.35 ppm. Experimental results from this study showed the -CH2- protons at 5.94 ppm when the ester was at the 5-position and at 5.42 ppm when the ester was at the 4-position. Based on these assignments, the major product in the reaction of ethyl phenylpropiolate and benzyl azide was determined to be ethyl 1-benzyl-4-phenyl-1,2,3-triazole-5-carboxylate (32% yield). Ethyl 1-benzyl-5-phenyl-1,2,3triazole-4-carboxylate was isolated at a significantly lower yield (14%). The sterics caused by the benzyl group apparently influenced the reaction to produce more of the 4-phenyl isomer than the 5-phenyl isomer (a ratio of about 2.3:1). The results obtained in this study reflect the conclusion obtained by Cwiklicki and Rehse in that the 4-phenyl triazole was the major product. Another interesting observation occurred in the reaction of 2-ethynylpyridine and benzyl azide (entry 4, Table 3). 2-Ethynylpyridine contained a nitrogen in the aromatic ring which caused the electronegativity of the ring to increase, and thereby also increased the electronegativity of the alkyne. This resulted in the need for a shorter overall reaction time and lower power settings on the microwave compared to the reaction of phenylacetylene and benzyl azide. 51 Although a few publications were found which included the reaction of 2ethynylpyridine and benzyl azide, most of them did not provide adequate spectral data for comparison. A group lead by Warren G. Lewis reported a triazole created using 2ethynylpyridine and benzyl azide, however the paper did not give any specific data for this particular triazole.104 Another literature report by Gonda and Novák105 cited the spectroscopic data for their study using bis-triphenylphosphano complexes of copper (I) carboxylates as efficient catalysts for synthesizing 1,2,3-triazoles. According to this literature source, the reaction utilizing 2-ethynylpyridine formed only the 4-pyridyl isomer. Using a domestic microwave, 2ethynylpyridine easily underwent reaction with benzyl azide to form a triazole without the use of any catalyst. The method employed showed that both the 4-pyridyl and 5-pyridyl-1,2,3-triazoles were formed. The isomers were separated by chromatography and then analyzed by 1H NMR (Figures 19 and 20). 52 SpinWorks 2.3: C Ph B CH2 N N N C A H D B D D N A D D D PPM 8.6 8.4 8.2 8.0 7.8 7.6 file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 4\2007\042707_A\1\fid expt: <zg30> transmitter freq.: 300.144690 MHz time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 32 7.4 7.2 7.0 6.8 6.6 6.4 6.2 6.0 freq. of 0 ppm: 300.142603 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 Figure 19. The 1H NMR of 1-benzyl-4-pyridyl-1,2,3-triazole in CDCl3. SpinWorks 2.3: C Ph B CH2 D D D N N N N D PPM 8.4 8.2 8.0 7.8 B H A C A D D D 7.6 file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 4\2007\042707_B\1\fid expt: <zg30> transmitter freq.: 300.144690 MHz time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 84 7.4 7.2 7.0 6.8 6.6 6.4 6.2 freq. of 0 ppm: 300.142603 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 Figure 20. The 1H NMR of 1-benzyl-5-pyridyl-1,2,3-triazole in CDCl3. 6.0 5.8 5.6 53 Initially, the 1H NMR spectra for each isomer were identified by using a pattern established by previously discussed triazoles, i.e. that when the proton was in the 4-postion of the triazole ring, the singlet observed is further downfield compared to that of the proton on the 5position in the other regioisomer. The benzyl group (-CH2Ph) also showed a typical pattern for chemical shifts in the two isomers. The -CH2- singlet for the benzyl group of the 5-pyridyl triazole was observed further upfield compared to the -CH2- singlet for the 4- pyridyl isomer. Even though these comparisons suggest the correct isomer identification, it was felt that these NMR trends were not enough to confirm the isomers’ identity. Also, Gonda’s literature only had spectral data for the 4-isomer triazole and did not describe the 5-isomer. From this, it was felt that spectral confirmation was needed. Nuclear Overhauser Effect (nOe) experiments were therefore performed to confirm the initial assignments. An nOe experiment is used to show which hydrogens interact with other neighboring hydrogen through space, since nOe effects become smaller as the atoms being studied get are farther apart in the 3-dimensional structure of the molecule. This effect can be seen as positive peaks observed for protons whose area has been enhanced by the nOe interaction, while negative peaks show no interaction. Two hydrogens appeared to be well placed for an nOe study: the benzyl -CH2- and the single hydrogen on the triazole ring. In the 4-substituted isomer, the hydrogens should be fairly close together, while the 5-substituted isomer then would be rather far apart. The nOe spectra were obtained for each isomer and are shown in Figures 21 and 22 below. 54 B TMS A Ph B CH2 N HA N N N Figure 21. nOe spectrum for 1-benzyl-4-pyridinyl-1,2,3-triazole in CDCl3. A B TMS Ph B CH2 N N N N H A Figure 22. nOe spectrum for 1-benzyl-5-pyridinyl-1,2,3-triazole in CDCl3. 55 In these studies, the triazole ring hydrogen (A) was irradiated and the benzyl -CH2protons (B) were observed for any changes. The 4-pyridyl isomer, in which the triazole hydrogen (A) was closer to the benzyl -CH2- protons (B), was observed to have a large positive nOe effect (Figure 21). For the 5-pyridyl isomer, in which the triazole hydrogen (A) was further from the benzyl -CH2- protons (B) the signal appeared to have a very small positive nOe effect (Figure 22) suggesting that these protons were too far apart to give a large nOe effect. While both of these nOe spectra also contain small negative peaks, these do not represent nOe effects for each proton. These negative peaks are considered to be false positive information and are obtained when the T1 time for the selected proton was not set properly during the set up for the nOe experiment. Overall, the nOe studies were conclusive in reinforcing the identification for each of the isomers isolated, i.e. that the 1-benzyl-5-pyridinyl-1,2,3-triazole was isolated with the highest isolated yield of 40.1% and 1-benzyl-4-pyridinyl-1,2,3-triazole was isolated with the lowest yield of 14%. The results obtained from the study of 2-ethynylpyridine and benzyl azide resulted in an unexpected outcome where the 5-pyridyl triazole dominated in yield compared to the 4-pyridyl isomer, since Gonda reported the use of a copper catalyst which resulted with the selective formation of the 4-pyridyl isomer. Gonda’s reaction, however, took place using a copper catalyst which has been known to influence the regioselectivity of these reactions. It is clear that in this microwave driven reaction steric factors did not dominate, since the more hindered 5-pyridyl isomer was formed in a 2.9:1 ratio over the less hindered 4-pyridyl isomer. Further studies are needed to better understand the observed selectivity. The final reaction studied using an unsymmetric alkyne was the reaction of propargyl alcohol with 2-(2-azidoethoxy)ethanol. This azide was synthesized and studied because it contained a second functional group (an alcohol) on the azide. Alcohols are important in organic 56 chemistry because they can be converted into many other types of functional groups using a variety of reactions. Alcohol groups are versatile and may be used as building blocks for larger, more complex molecules which may be used for a variety of applications ranging from food flavorings and fragrances to biological applications. The reaction of 2-(2-Azidoethoxy)ethanol and propargyl alcohol was accomplished successfully to produce 1-(2-(2-azidoethoxy)ethanol)-4hydroxymethyl-1,2,3-triazole with a yield of 36.1% and 1-(2-(2-azidoethoxy)ethanol)-5hydroxymethyl-1,2,3-triazole with a yield of 32.6% (entry 5, Table 3). The 1H NMR spectra of both regioisomers are shown below in Figures 23 and 24. 57 SpinWorks 2.3: C G SpinWorks 2.3: B, H F E A H A x D OH G O 7.6 7.4 4.2 4.0 3.8 3.6 3.4 H N Unknown Contaminent N HOH2C C B 7.8 1.000 4.4 N PPM E, F D 7.2 7.0 6.8 6.6 6.4 6.2 6.0 file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 4\2008\021308_144B\1\fid expt: <zg30> transmitter freq.: 300.144690 MHz time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 16 5.8 5.6 PPM 1 5.4 5.2 freq. of 0 ppm: 300.142596 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 7.6 5.0 4.8 4.6 7.2 4.4 6.8 4.2 4.0 6.4 3.8 6.0 3.6 3.4 5.6 5.2 4.8 Figure 23. The H NMR of 1-(2-(2-azidoethoxy)ethanol)-4-hydroxymethyl-1,2,3-triazole in CDCl3. SpinWorks 2.3: file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 4\2008\021308_144B\1\fid expt: <zg30> transmitter freq.: 300.144690 MHz time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 16 C B HOH2C F D OH G O x freq. of 0 ppm: 300.142 processed size: 32768 LB: 0.000 GB: 0.00 E, F B, C E 4.0 G H SpinWorks 2.3: 4.4 1.000 H 4.6 N 4.4 4.2 4.0 3.8 3.6 3.4 N A H A N D PPM 7.4 7.2 7.0 6.8 6.6 6.4 6.2 file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 4\2008\021408_146A\1\fid expt: <zg30> transmitter freq.: 300.144690 MHz time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 32 6.0 5.8 5.6 5.4 5.2 5.0 4.8 4.6 4.4 4.2 4.0 3.8 3.6 3.4 3.2 freq. of 0 ppm: 300.142599 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 Figure 24. The 1H NMR of 1-(2-(2-azidoethoxy)ethanol)-5-hydroxymethyl-1,2,3-triazole in PPM 7.2 6.8 6.4 6.0 5.6 5.2 CDCl3. file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 4\2008\021308_144A\1\fid expt: <zg30> transmitter freq.: 300.144690 MHz time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 16 4.8 4.4 freq. of 0 ppm processed siz LB: 0.000 58 The two isomers were separated via flash column chromatography and a D2O shake was used to determine the location of the alcohol groups in the 1H NMRs of both isomers. From the D2O exchange, the disappearances of peaks for the alcohol protons for each isomer were observed. Determining one isomer from the other cannot be achieved by locating where the alcohol protons are. Identifying which isomer corresponded to each 1H NMR spectum was accomplished by correlation to a synthesis published by Molteni.106 Molteni reported a triazole that was structurally similar to the triazoles synthesized in this study, however Molteni’s triazole contained a methoxy group on the end of the N-1 carbon chain rather than an alcohol. According to this literature reference, the lone hydrogen in the 4- or 5-isomer was used to assist in the determination of the absolute structure of the isomer. The proton in the 4- and 5- positions were reported at 8.11 ppm and 7.96 ppm, respectively, suggesting that the proton located at the 4position of the triazole ring was more deshielded than that of a hydrogen in the 5-position. Experimental results from this microwave study showed protons at 7.96 ppm and 7.61 ppm, were therefore assigned as the 4- and 5-positions, respectively. Using the same literature reference, the remaining protons were assigned by observing the similarities in chemical shifts between the triazole synthesized in this study and the one reported by Molteni. Identifying which alcohol peak was accomplished by observing where the alcohol for the hydroxymethyl was located. From the D2O exchange, the peaks for the alcohol protons for the 4hydroxymethyl isomer were determined to be at 3.5 ppm (C) and 5.2 ppm (D). For the 5hydroxymethyl isomer, the peaks for the alcohol protons were determined to be at 4.6 ppm (C) and 5.4 ppm (D). Proton (D) was easily distinguished in both spectra since it did not overlap with other signals. However, the alcohol protons from (C) were not as obvious. The 1H NMR spectra illustrating the transformations are shown below in Figures 25 and 26. 59 C D 2O SpinWorks 2.3: 4.00 3.90 PPM 3.80 3.70 4.00 3.60 3.50 Documents\Research\BOOK 4\2008\021308_144B\1\fid expt: <zg30> 3.903.40 3.80 3.30 3.20 3.70 3.10 3.60 3.00 2.90 3.50 3.30 3.20 3.10 freq. of 0 ppm: 300.142596 MHz processed size: 32768 complex points file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 4\2008\021408_146B_D2O\1\fid expt: <zg30> LB: 0.000 GB: 0.0000 transmitter freq.: 300.144690 MHz 1 time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 32 z/pt 3.40 freq. of 0 ppm: 300.142590 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 Figure 25. H NMR of 1-(2-(2-azidoethoxy)ethanol)-4-hydroxymethyl-1,2,3-triazole in CDCl3 showing the D2O exchange from alcohol to a deuterated alcohol. C D 2O 4.90 4.80 PPM 4.60 4.70 uments\Research\BOOK 4\2008\021308_144A\1\fid expt: <zg30> 4.74 4.50 4.72 4.70 4.40 4.68 4.66 4.30 4.64 freq. of 0C:\Documents ppm: 300.142597 MHzSettings\Cathleen Roush\My Documents\Research\BOOK 4\2008\021408_146A_D2O\1\fid file: and processed size: 32768 complex points transmitter freq.: 300.144690 MHz LB: 0.000 GB: 0.0000 1 time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 32 4.62 4.20 expt: <zg30> 4.60 4.58 4.56 4.54 4.52 freq. of 0 ppm: 300.142594 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 Figure 26. H NMR of 1-(2-(2-azidoethoxy)ethanol)-5-hydroxymethyl-1,2,3-triazole in CDCl3 showing the D2O exchange from alcohol to a deuterated alcohol. 4.50 4.48 60 With the observed disappearance of the alcohol peak for both isomers, identifying which alcohol peak belonged to the appropriate alcohol on each isomer. Using Molteni literature, the 4hydroxymethyl alcohol proton was determined to be at 3.5 ppm (C), leaving the 2-ethoxyethanol alcohol to be at 5.2 ppm (D). In the 5-hydroxymethyl isomer, the 2-ethoxyethanol was determined to be at 5.4 ppm (D), however the 5-hydroxymethyl alcohol proton was observed further downfield at 4.6 ppm (C) compared to the 4-isomer. With the hydroxymethyl in the 5position of the triazole, it is possibe that the alcohol can interact with the alcohol from the 2ethoxyethanol through intramolecular hydrogen bond which would result in a downfield chemical shift. The hydroxymethyl group on the alkyne was small, so it would not have been expected to exert a lot of steric influence for regioselectivity, as can be seen from the 1.1 : 1 ratio of isomers produced. Interestingly, the reaction proceeded with a very high yield (68.7 %) which might be attributed to the smaller steric hinderance in this system compared to previously discussed triazoles. 61 Synthesis of Bis-1,2,3-Triazoles Since the discovery and recent development of the “click” cycloaddition reaction, the 1,2,3-triazole heterocyclic motif has rapidly become one of the most popular structures in conjugate chemistry finding applications in the preparation of hybrid compounds, surface modification of materials and biomaterials, and molecular scaffolding.8, 50 Similarly to this breakthrough in monocyclic triazole chemistry, the 1,4-bis(azidomethyl)benzenes counterparts (Scheme 17) have become very popular to use in macromolecules,107 functionalized surfaces,108 multicomponent cascade reactions109 and to build libraries of various 1,2,3-triazoles rapidly and efficiently.110 The diverse applications for bis(azidomethyl)benzene made the microwave-driven synthesis of difunctional 1,2,3-triazoles an interesting subject for study. N N N N R R R N N N N N N N R R R N R N N N N N N R Scheme 17. Symmetrically substituted bis(1,2,3-triazoles). Various bis-triazoles might have been synthesized from any of the previously used alkynes in the studies already presented. However, careful consideration was taken to select an alkyne that would produce the best results. The alkynes chosen for the production of bis-triazoles 62 were selected from successful mono-triazole cyclization reactions accomplished when using benzyl azide. Literature sources were used as comparison for spectral data obtained in these reactions. The bis-triazoles were synthesized using 1,4-bis(azidomethyl)benzene in the reaction with three alkynes: dimethyl acetylenedicarboxylate, diphenylacetylene and ethyl propiolate. The results of these studies are summarized in Table 4. Table 4. Reactions between an alkyne and 1,4-bis(azidomethyl)benzene. Entry Azide Alkyne Reaction Conditions Products H3CH2CO2C N3 H3CO2C C 1 C CO2CH3 N3 Yield CO2CH2CH3 N N N 10% power/1 min 98% N N N H3CH2CO2C Ph N3 2 Ph N N N3 Ph C C Ph CO2CH2CH3 N 100% power/6 min 32% N N N Ph H3CH2CO2C N3 3 H N N H C N3 C CO2CH2CH3 30% power/5 min Ph N 35% N N N H CO2CH2CH3 63 64 It was anticipated that a reaction between an alkyne and a bis-azide would be most successful when the alkynes used were symmetric since the formation of and purification of isomeric product would then be avoided. The success observed in preliminary studies suggested that symmetric alkynes, when reacted with a bis-azide, would produce similar results. The alkynes chosen for this study were the most successful electron poor alkyne and electron rich alkyne. In the preliminary studies, dimethyl acetylenedicarboxylate and diphenylacetylene produced the highest yields of 98% and 80%, respectively, in the synthesis of single triazole rings. These results are summarized in Table 2, entries 2 & 3. A group lead by Sultan Abu-Orabi111 reported the formation of a triazole product from the reaction between 1,4-bis(azidomethyl)benzene and dimethyl acetylenedicarboxylate, obtaining product in 96% yield. According to this literature source, the reaction utilizing dimethyl acetylenedicarboxylate formed the bis-1,2,3-triazole by using conventional heating methods. Using a domestic microwave, dimethyl acetylenedicarboxylate easily underwent reaction with 1,4-bis(azidomethyl)benzene to obtain the bis-1,2,3-triazole without the use of any catalyst or added solvent. This microwave reaction successfully produced tetramethyl 1,1’-(p- phenylenedimethylene) bis[1H-1,2,3-triazole-4,5-dicarboxlate] (entry 1, Table 4) in a yield of 98%, and it was analyzed without further purification. The 1H NMR of this product is shown in Figure 27. 65 SpinWorks 2.3: N N A C D N H3CO2C CO2CH3 B A N D D A D PPM 8.0 C 7.0 6.0 file: F:\Research\BOOK 4\2008\081308_crude product\1\fid expt: <zg30> transmitter freq.: 300.144690 MHz time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 16 5.0 D B 4.0 CO2CH3 H3CO2C N N C B TMS acetone 3.0 2.0 1.0 0.0 freq. of 0 ppm: 300.142592 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 Figure 27. The 1H NMR of tetramethyl 1,1’-(p-phenylenedimethylene)bis [1H-1,2,3-triazole-4,5dicarboxlate] in CDCl3. The reaction conditions used with 1,4-bis(azidomethyl)benzene differed from that of benzyl azide when reacted with dimethylacetylene dicarboxylate. A preliminary study showed that benzyl azide reacted successfully with this alkyne at 30% power for 30 seconds. Using the same reaction conditions for 1,4-bis(azidomethyl)benzene and dimethyl acetylenedicarboxylate proved to be difficult to control, and the reaction overheated very quickly. The solution to this issue of overheating was to decrease the amount of applied power and to increase reaction times so as to slowly encourage the reaction to take place, heating at 10% power for a full minute. By applying less power the reaction was easily controlled with excellent results, providing the desired triazole product in 98% yield. In the 1H NMR spectrum (Figure 27), the four benzylic protons (C) and aromatic protons (D) in the product appeared as sharp singlets with chemical shifts of 5.79 ppm and 7.25, respectively. The methyl esters (A) and (B) appeared as two nearly overlapping singlets. The 66 methyl ester protons in the 5-position (A) were assigned to the peak observed at 3.96 ppm due to their proximity to the N-1 nitrogen. The methyl ester protons (B) found in the 4-position were assigned to the more upfield peak at 3.88 ppm. Another symmetric alkyne used in these bis-triazole studies was diphenylacetylene (entry 2, Table 4). Previous studies showed that phenylacetylene exhibited unusual reactivity with benzyl azide under these conditions to form a diphenyl triazole, and from this, the reaction of diphenylacetylene and 1,4-bis(azidomethyl) benzene was observed for any similarities to the previous reaction. Had both triazole cyclization reactions proceeded at an 80% yield, the bistriazole would have been expected to form in a 64% isolated yield (80% x 80%). The initial reaction between 1,4-bis(azidomethyl)benzene and diphenylacetylene was attempted under the same conditions as the reaction with the mono-azide. Using the initial reaction conditions of 80% power for 15 minutes, the mixture did not show major color changes from a yellow liquid of the original mixture. Analysis of the crude product at this point indicated that much of the phenylacetylene had not reacted with the 1,4-bis(azidomethyl)benzene, even after extended heating. The reaction was attempted again, raising the irradiation power from 80% to 100%, and heated the mixture until there was a visible color change from pale yellow to a deep, amber red (~ 6 minutes). The crude mixture was purified by recrystallization utilizing a solvent mixture of petroleum ether and methanol. The purified triazole was isolated as a dark brown solid (32%). The 1H NMR of tetraphenyl 1,1’-(p-phenylenedimethylene)bis [4,5-diphenyl-1H-1,2,3-triazole] is shown in Figure 28. The 1H NMR spectrum shows that no remaining starting material was present. The single peak for the -CH2- (F) and (G) had an upfield chemical shift of 4.33 ppm to 5.35 ppm. Integrations of these peaks show that peaks for benzyl protons (F) and (G) integrate to 4 protons. The aromatic region of the spectrum contains many peaks which were found to integrate to 24 aromatic protons. 67 SpinWorks 2.3: B A N B N N N B Ph B Ph B B B Ph B Ph A B N N CHCl3 PPM 7.6 7.4 7.2 7.0 6.8 file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 5\2008\100908_CR15\1\fid expt: <zg30> transmitter freq.: 300.144690 MHz time domain size: 65536 points width: 6172.84 Hz = 20.566213 ppm = 0.094190 Hz/pt number of scans: 32 A 6.6 6.4 6.2 6.0 5.8 5.6 5.4 freq. of 0 ppm: 300.142606 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 Figure 28. The 1H NMR of tetraphenyl 1,1’-(p-phenylenedimethylene)bis [4,5-diphenyl-1H1,2,3-triazole] in CDCl3. Analysis of the crude did afford the desired bis-triazole product, however the isolated yield dropped to 32%, compared to the reaction of diphenyl acetylene and benzyl azide which produced 80% of the mono-triazole. This drop in yield could be due to the overall increase in sterics for the product tetraphenyl 1,1’-(p-phenylenedimethylene) bis[4,5-diphenyl-1H-1,2,3triazole]. With this reaction, the bis-triazole now contained four appended phenyl groups causing the space within this molecule to become crowded. This crowding might have affected the rate of the reaction and slowed it down, making it difficult to achieve higher yield for the bis-1,2,3triazole with the current designed method. This study was extended to include the reaction of a lower polarity alkyne (ethyl propiolate) and 1,4-bis(azidomethyl)benzene (entry 3, Table 4). Using ethyl propiolate was of interest in order to observe the success of a reaction when utilizing an unsymmetric alkyne. This would increase the complexity of purification for every isomer produced in this reaction, since 68 each triazole ring formed has the potential to exist in two regioisomeric forms. The crude product from the reaction was purified by recrystallization from petroleum ether and methanol to give a fluffy, bright yellow solid with an isolated yield of 35%. The 1H NMR of diethyl 1,1’-(pphenylenedimethylene)bis [1H-1,2,3-triazole-4-carboxylate] is shown in Figure 29. SpinWorks 2.3: proton D E H3C C O H2 C O H C A A N N N N N B N B C A A C H2 O H 7.6 7.2 6.8 6.4 6.0 5.6 file: C:\Documents and Settings\Cathleen Roush\My Documents\Research\BOOK 5\2011\EPBBA\1\fid expt: <zg30> transmitter freq.: 300.131853 MHz time domain size: 65536 points width: 6218.91 Hz = 20.720578 ppm = 0.094893 Hz/pt number of scans: 128 5.2 4.8 4.4 4.0 3.6 3.2 TMS E D CDCl3 C PPM Unknown Contaminant B 2.8 2.4 2.0 1.6 CH3 D C A E O 1.2 0.8 0.4 -0.0 freq. of 0 ppm: 300.130005 MHz processed size: 32768 complex points LB: 0.000 GB: 0.0000 Figure 29. The 1H NMR of diethyl 1,1’-(p-phenylenedimethylene)bis[1H-1,2,3-triazole-4carboxylate] in CDCl3. The 1H NMR appeared very simple, but revealed a lot about the product isolated. The unknown contaminant at 3.94 ppm could not be identified as starting material or as any known solvent impurities. Further spectral analysis did not place this peak as part of the product molecule, since this peak did not appear in the crude product prior to recrystallization. Thus, this contaminant must have entered the system during purification process. 69 The product was expected to exist in isomeric forms, however the 1H NMR showed what appeared to be a single product isomer. In great contrast to the mono-cyclization reactions conducted with benzyl azide, the reaction of ethyl propiolate and 1,4-bis(azidomethyl)benzene displayed complete regiospecificity under similar conditions since only one product was obtained after purification. The success of producing a single product was highly unexpected and the analysis of the spectrum was confirmed by comparison to literature values.111 Its possible that complete regiospecificity was accomplished due to the influence of steric effects. The molecule was slightly crowded with the two 1,2,3-triazoles on the para positions of the benzene ring. However, the sterics in the molecule was reduced when the two ester groups on each triazoles are positioned as far away from each other as possible, i.e. in the 4-position of each triazole. 70 Other Attempted Reactions The reaction of propiolic acid and 1,4-bis(azidomethyl) benzene was attempted in order to explore the possibility of a double decarboxylation reaction in the formation of a bis-triazole. Initial studies began with propiolic acid (Figure 30) and 1,4-bis(azidomethyl) benzene placed in an 25mL Erlenmeyer flask. However, in a matter of seconds, a visible color change from clear to a deep, amber red was observed even in the absence of any external heat source. The flask was placed in the microwave for 30% power for 3 minutes for additional heating to ensure completeness of the reaction. The resulting crude product showed that while some product was formed, starting materials still remained. The reaction was attempted once more, and the flask was irradiated at 50% power for 2 minutes. The crude product was analyzed immediately, but the results were identical to the first experiment. The 1H NMR showed that some product had formed, but the spectrum appeared to contain other unknown peaks that could not be identified. Since the ability to control this reaction to form a clean product could not be accomplished over multiple attempts, it was not pursued further. O H C C Propiolic Acid N3 OH N3 1,4-bis(azidomethyl)benzene Figure 30. Alkynes used for attempted reactions. 71 The success of this study forming various triazoles using a domestic microwave lead to the question of how these reactions might perform in a laboratory grade microwave. Therefore, several reactions were attempted in the MARS laboratory microwave, owned by the Department of Chemistry at California State University, Sacramento (CSUS), which were successful using the domestic microwave oven to optimize the comparisons. The reactions attempted utilized propiolic acid, diphenylacetylene and dimethyl acetylenedicarboxylate, each reacting with benzyl azide. In addition a unique reaction was attempted, that of acetylenedicarboxamide and benzyl azide. In every case, the reactions performed produced very little product. Since the MARS microwave system works better at larger scales and azides pose a potential explosion hazard when heated in the microwave, the reactions could not be scaled up to properly be optimized in the MARS microwave oven. It is also important to note that while no explosions occurred in the initial attempts to utilize the MARS instrument with these organic azides, care was given whenever azides were used to avoid potential hazards. 72 Chapter 4 CONCLUSIONS Substituted 1,2,3-triazoles can be synthesized from a variety of organic azides and alkynes via a simple cycloaddition reaction under neat conditions. The method designed for the synthesis of 1,2,3-triazole made is possible to synthesize various types of triazoles. Through the use of standardized glassware, the overall cost for the reactions dropped dramatically, making it attractive to synthesize many compounds since there was no need for specialized, expensive glassware. The reactions performed in this study illustrated several of the guiding principles of Green Chemistry, namely that a reaction could be performed with no solvent, no catalyst, short reactions and easy clean-ups. This study also highlighted the benefits in utilizing a microwave oven for organic synthesis. Using a microwave oven allowed for reactions to be performed more efficiently, affording reduced reaction times, enhanced yields, and selectivity. In the absence of a catalyst, moderate to high yields of 1,2,3-triazoles were seen from both electron-rich and electron-poor alkynes upon heating with an azide in a domestic microwave oven. This method was also applied to creating larger, more complex bis-triazoles. These bis-triazoles formed were synthesized using 1,4-bis(azidomethyl)benzene and three alkynes: dimethyl acetylenedicarboxylate, diphenylacetylene and ethyl propiolate, and afforded product in low to high yields. Thus, this simple, green, microwave-assisted synthesis provides an effective approach to the synthesis of a large variety of substituted 1,2,3-triazoles. The development of a novel microwave enhanced synthetic protocol for the formation of 1,2,3-triazole derivatives has been accomplished. This eco-friendly, solvent-free approach using microwave irradiation gives many possibilities for conducting rapid 1,2,3-triazole synthesis. Triazoles have important properties and the potential to be incorporated into so many useful 73 bioactive compounds, making them of intense interest. The success of these reactions highlights the need for future work in this area, which should include: Performing reactions in a laboratory grade microwave for comparative results Synthesizing 1,2,3-triazoles using various other azides (i.e. azidomethyl phenyl sulfide) and multi-azide starting material (i.e. Polyoxyethylene bis(azide)) Synthesizing fused 1,2,3-triazole rings Computational studies for the formation of simple and complex 1,2,3-triazoles 74 Chapter 5 EXPERIMENTAL General Information Abbreviations. Dimethyl sulfoxide (DMSO); Dimethyl Formamide (DMF); Deuteratated chloroform (CDCl3); Deuterated-d6 Dimethyl sulfoxide (d6-DMSO). Spectral. All spectra were obtained from a 300 MHz Bruker Avance AC 300 NMR spectrometer in either CDCl3 or d6-DMSO. Materials. Sodium azide was purchased from Matheson Coleman & Bell. Substrates purchased from Acros Organics were benzyl chloride, ethyl bromoacetate, 2-(2-chloroethoxy)ethanol, 1,4bis(bromomethyl)benzene, ethyl propiolate, dimethylacetylene dicarboxylate, ethylphenyl propiolate, diphenylacetylene, propiolic acid, phenyl acetylene and phenyl propiolic acd. Substrates purchased from Aldrich Chemical Company, Inc. were azidotrimethylsilane, 2-ethynyl pyridine and propargyl alcohol. All other reagents and solvents were of analytical grade, were purchases from local suppliers and were used as obtained. Overall synthesis. Synthesis for 1,2,3-triazoles were conducted in an Emerson Model 8912B microwave oven (900 Watts). The synthesis for benzyl azide, ethyl 2-azidoacetate, 2-(2azidoethoxy)ethanol and 1,4-bis(azidomethyl)benzene were not synthesized using the microwave oven, but by conventional methods. 75 Synthesis of the Azide Starting Materials Benzyl azide (see Tables 2 & 3)112 A mixture of benzyl chloride (23 mL, 0.20 mol) and two equivalents of sodium azide (25.699 g, 0.395 mols) were placed in a 125 mL Erlenmeyer flask with 45 mL of DMSO. The flask was loosely corked and was allowed to stir overnight. The mixture was then diluted with 80 mL anhydrous diethyl ether and extracted with water (3 x 200 mL). The organic layer was dried over sodium sulfate and concentrated by rotary evaporation. The product was isolated as a pale yellow liquid (24.85 g, 0.187 mol, 93%). 300 MHz 1H NMR in CDCl3 (δ ppm): 4.31 (s, 2H); 7.31-7.45 (m, 5H). 75 MHz 13C NMR in CDCl3 (δ ppm): 54.5; 128.0; 128.1; 128.6; 135.3. Ethyl 2-azidoacetate (see Table 2)113 Ethyl bromoacetate (6.60 mL, 0.0595 mol) and sodium azide (7.79 g, 0.119 mol) were placed in a 100 mL round-bottomed flask with 50 mL DMSO. The flask was loosely covered and warmed at 55 oC for 2 h with stirring. The mixture was then diluted with 100 mL anhydrous diethyl ether and extracted with water (4 x 125 mL). The combined organic layer was washed with brine (2 x 50 mL), dried over anhydrous sodium sulfate, filtered and concentrated by rotary evaporation leaving a colorless liquid (6.84 g, 0.0529 mol, 89%). 300 MHz 1H NMR in CDCl3 (δ ppm): 1.31 (t, 3H, -CH3, J = 7.13 Hz); 3.97 (s, 2H, - CH2-C=O); 4.27 (q, 2H, -CH2-, J = 7.20 Hz). 75 MHz 13C NMR in CDCl3 (δ ppm): 13.7; 50.0; 61.5; 168.2. 76 2-(2-Azidoethoxy)ethanol (see Table 3)114 NaN3 (4.50 g, 0.070 mol), tetrabutylammonium iodide (2.50 g, 0.060 mol), and 18-crown-6 (14 mg, 0.0530 mmol) were added to a solution of 2-(2-chloroethoxy)ethanol (5 mL, 0.045 mmol) in 2-butanone (25 mL, 0.0280 mol). The mixture was refluxed at 90 °C for 2 days with stirring. The resulting precipitate was removed by filtration and rinsed with acetone. The combined organic solutions were concentrated via rotary evaporation and purified by distillation, leaving a pale yellow liquid (4.65 g, 0.035 mol, 75%). Results obtained agreed well with literature. 300 MHz 1H NMR in CDCl3 (δ ppm): 3.73-3.80 (t, 2H, -CH2-O-, J = 4.6 Hz), 3.67-3.73 (t, 2H, N3-CH2-, J = 5.0 Hz), 3.58-3.65 (t, 2H, -O-CH2-, J = 4.5 Hz), 3.38-3.46 (t, 2H, -CH2-OH, J = 4.9 Hz), 2.58 (s, 1H, OH). 75 MHz 13C NMR in CDCl3 (δ ppm): 54.9; 61.3; 69.7; 72.3. 1,4-bis(azidomethyl)benzene (see Table 4)115 1,4-bis(Bromomethyl)benzene (10.0 g, 0.038 mol) and sodium azide (5.0 g, 0.077 mol) were placed in a 100 mL round-bottomed flask with 25 mL of DMF. The mixture was refluxed overnight at 65 °C with stirring. The mixture was then diluted with 200 mL of water and extracted with anhydrous diethyl ether (3 x 100 mL). The combined organic layers were washed with brine (3 x 100 mL), dried over anhydrous sodium sulfate, filtered and concentrated by rotary evaporation leaving a colorless liquid (6.62 g, 0.035 mol, 93%). 300 MHz 1H NMR in CDCl3 (δ ppm): 4.33 (s, 4H); 7.33 (s, 4H). 75 MHz 13C NMR in CDCl3 (δ ppm): 54.0; 128.3; 135.4. 77 Mono-1,2,3-Triazoles 1-Benzyl-1,2,3-triazole (Table 2, entry 1) 116 A mixture of propiolic acid (0.400 mL, 6.51 mmols) and benzyl azide (0.579 g, 4.34 mmols) were placed in a 25 mL Erlenmeyer flask and loosely covered with a watch glass. The Erlenmeyer flask was placed in a microwave and irradiated at 30 % power for 5 minutes. The flask was allowed to cool to room temperature, leaving a viscous brown oil which solidified overnight, mp 50-52 oC (0.687 g, 0.431 mmols, 99%). Results obtained agreed well with literature. 300 MHz 1H NMR (δ ppm): 5.57 (s, 2H); 7.23 – 7.30 (m, 2H); 7.34 – 7.41 (m, 3H); 7.47 (d, 1H, H-triazole, J = 0.75 Hz); 7.71 (d, 1H, H-triazole, J = 0.76 Hz). 75 MHz 13C NMR in CDCl3 (δ ppm): 53.22; 122.56; 127.23; 127.96; 128.34; 133.42; 133.90. 1-Benzyl-4,5-diphenyl-1,2,3-triazole (Table 2, entry 2) 52 A mixture of diphenylacetylene (0.852 g, 4.77 mmols) and benzyl azide (0.578 g, 4.34 mmols) were combined in a 25 mL Erlenmeyer flask and loosely covered with a watch glass. The Erlenmeyer flask was placed in a microwave and was irradiated at 80 % power for 15 minutes. The flask was allowed to cool to room temperature, leaving a viscous, red-brown oil which solidified overnight. The compound was analyzed without further purification, mp 109-112oC (1.06 g, 3.39 mmols, 80%). 300 MHz 1H NMR in CDCl3 (δ ppm): 5.40 (s, 2H); 6.99-7.05 (m, 2H); 7.11-7.17 (m, 2H); 7.227.27 (m, 5H); 7.34-7.47 (m, 4H); 7.52-7.58 (m, 2H). 75 MHz 13 C NMR in CDCl3 (δ ppm): 78 52.02; 126.68; 127.45; 127.67; 127.86; 128.10; 128.29; 128.39; 128.65; 129.11; 129.62; 130.08; 130.91; 131.58; 133.83; 135.34; 144.50. Dimethyl-1-benzyl-1,2,3-triazole-4,5-carboxylate (Table 2, entry 3) 117 A mixture of dimethyl acetylenedicarboxylate (0.530 mL, 4.34 mmols) and benzyl azide (0.580 g, 4.34 mmols) were combined in a 25 mL Erlenmeyer flask and loosely covered with a stopper. The Erlenmeyer flask was placed in a microwave and was irradiated at 30 % power for 30 seconds. The flask was allowed to cool to room temperature, leaving behind a thick, red oil. The product was analyzed without further purification (1.18 g, 0.49 mol, 98%). 300 MHz 1H NMR in CDCl3 (δ ppm): 3.86(s, 3H); 3.94 (s, 3H); 5.79 (s, 2H); 7.23-7.28 (m, 2H); 7.29-7.36 (m, 3H). 75 MHz 13 C NMR in CDCl3 (δ ppm): 52.54; 53.17; 53.79; 127.87; 128.70; 128.80; 129.67; 133.81; 140.07; 158.67; 160.29. Dimethyl-1-((carboethoxy)methyl)-1,2,3-triazole-4,5-dicarboxylate (Table 2, entry 4) A mixture of ethyl 2-azidoacetate (0.520 mL, 4.34 mmols) and 118 dimethyl acetylenedicarboxylate (0.561 g, 4.34 mmols) were combined in a 25 mL Erlenmeyer flask, loosely covered with a watch glass, placed in a microwave, and irradiated at 30 % power for 10 seconds. The flask was allowed to cool to room temperature, leaving a visous, red-brown oil, which solidified upon cooling. The product was then recrystallized with toluene, producing pale yellow needle crystals, mp 124-125oC (1.00 g, 3.70 mmols, 87%). 79 300 MHz 1H NMR in CDCl3 (δ ppm): 1.29 (t, 3H, -CH3, J = 7.2 Hz); 3.97 (s, 3H, O=C-CH3); 3.99 (s, 3H, O=C-CH3); 4.26 (q, 2H, -CH2-, J = 7.1 Hz); 5.45 (s, 2H, N-CH2-). 75 MHz 13C NMR in CDCl3 (δ ppm): 13.05; 50.64; 51.71; 52.39; 61.55; 128.91; 139.28; 157.61; 159.22; 164.48. Dimethyl-1H-1,2,3-triazole-4,5-carboxylate (Table 2, entry 5) 101 A mixture of dimethyl acetylenedicarboxylate (0.800 mL, 6.51 mmols) and azidotrimethylsilane (0.865 g, 6.51 mmols) were combined in a 25 mL Erlenmeyer flask, loosely covered with a stopper, placed in a microwave, and irradiated at 30 % power for 6 minutes. The flask was allowed to cool to room temperature. The crude product was dissolved in anhydrous diethyl ether and washed with sodium bicarbonate (1 x 25 mL) and brine (1 x 20 mL). The organic layer was dried over sodium sulfate, filtered and concentrated by rotary evaporation to leave a yellow solid, mp 125-128oC (0.736 g, 3.98 mmols, 61%). 300 MHz 1H NMR in d6-DMSO (δ ppm): 3.95(s, 6H); 16.28 (very broad s, 1H). 75 MHz 13C NMR in CDCl3 (δ ppm): 51.15; 136.60; 158.37. 1-Benzyl-4-phenyl-1,2,3-triazole & 1-Benzyl-5-phenyl-1,2,3-triazole (Table 4, entry 1) 52 A mixture of phenylacetylene (0.470 mL, 4.34 mmols) and benzyl azide (1.16 g, 4.34 mmols) were placed in a 25 mL Erlenmeyer flask and loosely covered with a watch glass. The Erlenmeyer flask was placed in a microwave and was irradiated at 80 % power for 9 minutes. The flask was allowed to cool to room temperature, and the crude product was purified by flash chromatography on silica gel using 100:1 dichloromethane-methanol as eluent, giving two 80 isomeric products. The 4-phenyl isomer was isolated as a pale yellow solid, mp 60-62 oC (0.244 g, 1.04 mmol, 24.2 %) and the 5-phenyl isomer was isolated as a pale yellow solid, mp 133-135 o C (0.242 g, 1.03 mmol, 24.1%). (4-phenyl) 300 MHz 1H NMR (δ ppm): 5.58 (s, 2H); 7.27 – 7.35 (m, 5H); 7.35 – 7.43 (m, 3H); 7.66(s, 1H, H-triazole); 7.76-7.83 (m, 2H). 75 MHz 13 C NMR in CDCl3 (δ ppm): 54.20; 119.44; 125.58; 127.94; 128.03; 128.65; 128.67; 129.04; 130.42; 134.56; 148.12. (5-phenyl) 300 MHz 1H NMR (δ ppm): 5.53 (s, 2H); 7.04 – 7.08 (m, 2H); 7.22-7.30 (m, 5H); 7.37-7.46 (m, 3H); 7.73 (s, 1H, H-triazole). 75 MHz 13C NMR in CDCl3 (δ ppm): 51.73; 126.76; 127.03; 128.03; 128.70; 128.75; 128.84; 129.40; 133.12; 135.38; 138.05. 1-Benzyl-5-phenyl-1,2,3-triazole & 1-Benzyl-4-phenyl-1,2,3-triazole (Table 4, entry 2) 1, 52, 119 A mixture of phenyl propiolic acid (0.634 g, 4.34 mmols) and benzyl azide (1.17 g, 8.68 mmols) were combined in a 25 mL Erlenmeyer flask and loosely covered with a watch glass. The Erlenmeyer flask was placed in a microwave and was irradiated at 30 % power for 6 minutes. After heating, the flask was allowed to cool to room temperature, and the crude product was purified by flash chromatography on silica gel using 1:100 methanol-dichloromethane, giving two isomeric products. The 4-phenyl isomer was isolated as a yellow solid, mp 62-64 oC (0.323 g, 1.38 mmols, 32 %) and the 5-phenyl isomer was isolated as yellow solid mp 135-136 oC (0.141 g, 0.599 mmols, 14%). (4-phenyl) 300 MHz 1H NMR (δ ppm): 5.57 (s, 2H); 7.27-7.34 (m, 5H); 7.34-7.42 (m, 3H); 7.65 (s, 1H, H-triazole); 7.77-7.82 (m, 2H). 75 MHz 13C NMR in CDCl3 (δ ppm): 54.20; 119.44; 125.58; 127.93; 128.03; 128.64; 128.68; 129.02; 130.45; 134.60; 148.09. 81 (5-phenyl) 300 MHz 1H NMR (δ ppm): 5.55 (s, 2H); 7.06 – 7.10 (m, 2H); 7.23-7.31 (m, 5H); 7.38 – 7.45 (m, 3H); 7.74 (s, 1H, H-triazole). 75 MHz 13C NMR in CDCl3 (δ ppm): 51.75; 126.79; 127.01; 128.02; 128.67; 128.75; 128.80; 129.35; 133.14; 135.38; 138.01. Ethyl 1-benzyl-4-phenyl-1,2,3-triazole-5-carboxylate & Ethyl 1-benzyl-5-phenyl-1,2,3-triazole4-carboxylate (Table 4, entry 3) 103 A mixture of ethyl phenylpropiolate (0.730 mL, 4.34 mmols) and benzyl azide (1.170 g, 8.68 mmols) were placed in a 25 mL Erlenmeyer flask and loosely covered with a watch glass. The Erlenmeyer flask was placed in a microwave and was irradiated at 30 % power for 6 minutes. The flask was allowed to cool to room temperature, and the crude product was purified by flash chromatography on silica gel using 1:100 methanol-dichloromethane as eluent, giving two isomeric products. The 4-phenyl isomer was isolated as a white solid, mp 97-99 oC (0.323 g, 1.05 mmols, 32 %) and the 5-phenyl isomer was isolated as a pale yellow oil, (0.141 g, 0.459 mmols, 14%). (4-phenyl) 300 MHz 1H NMR (δ ppm): 1.17 (t, 3H, CH3 on ethyl ester, J = 7.2 Hz); 4.42 (q, 2H, -CH2- on ethyl ester, J = 7.2 Hz); 5.94 (s, 2H, N-CH2-); 7.24 – 7.35 (m, 5H); 7.39 – 7.44 (m, 3H); 7.67 – 7.74 (m, 2H). 75 MHz 13C NMR in CDCl3 (δ ppm): 14.16; 52.20; 61.11; 125.98; 127.50; 128.39; 128.50; 128.78; 129.77; 130.07; 134.66; 137.16; 141.27; 160.90. (5-phenyl) 300 MHz 1H NMR (δ ppm): 1.25 (t, 3H, CH3 on ethyl ester, J = 7.0 Hz); 4.29 (q, 2H, -CH2- on ethyl ester, J = 7.1 Hz); 5.43 (s, 2H, N-CH2-); 6.95 – 7.03 (m, 2H); 7.16 – 7.22 (m, 2H); 7.22 – 7.29 (m, 3); 7.39 – 7.53 (m, 3H). 75 MHz 13C NMR in CDCl3 (δ ppm): 82 13.79; 54.24; 61.85; 124.13; 127.86; 127.97; 128.37; 128.78; 128.95; 129.44; 130.31; 135.26; 150.48; 159.17. 1-Benzyl-4-pyridinyl-1,2,3-triazole & 1-Benzyl-5-pyridinyl-1,2,3-triazole (Table 4, entry 4) 105 A mixture of 2-ethynylpyridine (1.75 mL, 1.74 mmols) and benzyl azide (0.464 g, 3.48 mmols) were placed in a 25 mL Erlenmeyer flask and loosely covered with a watch glass. The Erlenmeyer flask was placed in a microwave and was irradiated at 50 % power for 9 minutes. The flask was allowed to cool to room temperature, and the crude product was purified by flash chromatography on silica gel using 70:30 ethyl acetate-hexane as eluent, giving two isomeric products. The 4-pyridyl isomer was isolated as a white solid, mp 83-84 oC (0.164 g, 0.694 mmols, 40.1 %) and the 5-pyridyl isomer was isolated as an off-white solid, mp 105-107oC (0.0571 g, 0.242 mmols, 14.0%). (4-pyr) 300 MHz 1H NMR δ ppm): 6.16 (s, 2H); 7.20 - 7.30 (m, 6H); 7.51 - 7.56 (m, 1H); 7.70 - 7.76 (m, 1H); 8.00 (s, 1H, H-triazole); 8.67 - 8.71 (m, 1H). 75 MHz 13C NMR in CDCl3 (δ ppm): 53.09; 122.77; 123.31; 127.83; 127.86; 128.51; 133.68; 135.60; 136.13; 137.03; 147.07; 149.53. (5-pyr) 300 MHz 1H NMR (δ ppm): 5.89 (s, 2H); 7.20 - 7.24 (m, 1H); 7.31- 7.40 (m, 5H); 7.74 - 7.80 (m, 1H); 8.07 (s, 1H, H-triazole); 8.16 - 8.20 (m, 1H); 8.52 - 8.55 (m, 1H). 75 MHz 13C NMR in CDCl3 (δ ppm): 53.67; 119.49; 120.54; 121.20; 122.09; 127.54; 128.09; 128.41; 134.34; 136.15; 148.56; 150.26. 83 1-(2-(2-Azidoethoxy)ethanol)-4-hydroxymethyl-1,2,3-triazole & 1-(2-(2-Azidoethoxy) ethanol)5-hydroxymethyl-1,2,3-triazole (Table 4, entry 5)106 A mixture of propargyl alcohol (0.253 mL, 4.34 mmols) and 2-(2-Azidoethoxy) ethanol (0.570 g, 4.34 mmols) were placed in a 25 mL Erlenmeyer flask and loosely covered with a watch glass. The Erlenmeyer flask was placed in a microwave and was irradiated at 30% power for 6 minutes. The flask was allowed to cool to room temperature, and the crude product was purified by flash chromatography on silica gel using 80:20 diethyl ether-methanol as eluent, giving two isomeric products. The 4-hydroxymethyl isomer was isolated as a colorless thick oil (0.290 g, 1.55 mmol, 36.1%) and the 5-hydroxymethyl isomer was isolated as a colorless thick oil (0.262 g, 1.40 mmol, 32.6%). (4-hydroxymethyl) 300 MHz 1H NMR in DMSO-d6 (δ ppm): 3.38-3.53 (m, 5H); 3.763.85 (t, 2H, J = 5.3 Hz); 4.47-4.56 (m, 4H); 5.17-5.24 (t, 1H, -OH ether, J = 5.5 Hz); 7.96 (s, 1H, H-triazole). 75 MHz 13C NMR in DMSO-d6 (δ ppm): 49.50; 55.26; 60.32; 68.96; 72.23; 123.24; 147.97. (5-hydroxymethyl) 300 MHz 1H NMR in DMSO-d6 (δ ppm): 3.32-3.51 (m, 4H); 3.753.86 (t, 2H, J = 5.4 Hz); 4.44-4.57 (t, 2H, J = 5.5 Hz); 6.62(s, 2H); 4.64 (s, 1H, -OH ether); 5.395.49 (t, 1H, -OH, J = 5.7); 7.61 (s, 1H, H-triazole). 75 MHz 13C NMR in DMSO-d6 (δ ppm): 47.65; 52.11; 60.24; 69.26; 72.32; 132.25; 138.15. 84 Bis-1,2,3-Triazoles Tetramethyl 1,1’-(p-phenylenedimethylene)bis[1H-1,2,3-triazole-4,5-dicarboxlate] (Table 5, entry 1) 111 A mixture of dimethyl acetylenedicarboxylate (0.435 mL, 3.54 mmols) and 1,4bis(azidomethyl)benzene (0.399 g, 2.13 mmols) were combined in a 25 mL Erlenmeyer flask and loosely covered with a watch glass. The Erlenmeyer flask was placed in a microwave and was irradiated at 10 % power for 1 minute. The flask was allowed to cool to room temperature, leaving a viscous, yellow-brown oil which solidified overnight. The compound was analyzed without further purification, mp 141-142 oC (0.820 g, 1.74 mmols, 98%). 300 MHz 1H NMR in CDCl3 (δ ppm): 3.88 (s, 6H, -CH3 on ester); 3.96 (s, 6H-CH3 on ester); 5.79 (s, 4H, N-CH2-); 7.25 (s, 4H, ph). 75 MHz 13 C NMR in CDCl3 (δ ppm): 52.74; 53.37; 128.72; 134.80; 140.41; 158.72; 160.40. Tetraphenyl 1,1’-(p-phenylenedimethylene)bis[4,5-diphenyl-1H-1,2,3-triazole] (Table 5, entry 2) A mixture of diphenylacetylene (0.775 g, 4.34 mmols) and 1,4-bis(azidomethyl) benzene (0.815 g, 4.34 mmols) were combined in a 25 mL Erlenmeyer flask and loosely covered with a watch glass. The Erlenmeyer flask was placed in a microwave and was irradiated at 100 % power for 6 minutes. The flask was allowed to cool to room temperature, leaving a viscous, red oil which solidified overnight. The product was then recrystallized with methanol-petroleum ether, producing dark brown solid, mp 112-114 oC (0.750 g, 1.38 mmols, 32%). 85 300 MHz 1H NMR in CDCl3 (δ ppm): 5.35 (s, 4H, N-CH2-); 7.03-7.61 (m, 24H). 75 MHz 13C NMR in CDCl3 (δ ppm): 50.39; 128.33; 128.83; 128.99; 129.35; 133.44; 133.89; 145.51; 145.89. Diethyl 1,1’-(p-phenylenedimethylene)bis[1H-1,2,3-triazole-4-carboxylate] (Table 5, entry 3) 111 A mixture of ethylpropiolate (0.530 mL, 5.21 mmols) and 1,4-bis(azidomethyl) benzene (0.816 g, 4.34 mmols) were combined in a 25 mL Erlenmeyer flask and loosely covered with a watch glass. The Erlenmeyer flask was placed in a microwave and was irradiated at 50 % power for 5 minutes. After heating, the flask was allowed to cool to room temperature, and the resulting solid was then recrystallized with methanol-petroleum ether, producing a fluffy, bright yellow solid, mp 215-218 oC (0.492 g, 1.28 mmols, 35%). 300 MHz 1H NMR (δ ppm): 1.34-1.37 (t, 6H, -CH3 on ester, J=7.1 Hz); 4.35-4.45 (q, 4H, -CH2on ester, J=7.2 Hz); 5.59 (s, 4H, N-CH2-); 7.32 (s, 4H, ph); 8.00 (s, 2H, H-triazole). 75 MHz 13C NMR in CDCl3 (δ ppm): 14.34; 42.37; 52.38; 61.48; 91.92; 127.36; 129.22; 165.34. 86 REFERENCES 1. Mayot, E.; Gerardin-Charbonnier, C.; Selve, C., J. Fluor. Chem. 2005, 126, 715. 2. Borello, E.; Zecchina, A.; Guglielminotti, E., J. Chem. Soc. [Section] B: Organic 1969, 3, 307. 3. Gold, H., Jus. Lieb. Ann. Chem. 1965, 688, 205. 4. Katritzky, A. R.; Boulton, A. J., Advances in Heterocyclic Chemistry. Academic Press, Inc.: New York, 1974; Vol. 16, p 63-64. 5. Roumestant, M. L.; Viallefont, P.; Elguero, J.; Jacquier, R.; Arnal, E., Tet. Lett. 1969, 6, 495. 6. Stiefvater, O. L.; Jones, H.; Sheridan, J., Spec. Acta, Part A: Mol. Biom. Spec. 1970, 26, (4), 825. 7. Eicher, T.; Hauptmann, S.; Speicher, A., The Chemistry of Heterocylces: Structure, Reactions, Syntheses, and Applications. 2nd ed.; WILEY-VCH GmbH & Co. KGaA: Weinheim, 2003; p 200. 8. Kolb, H. C.; Sharpless, K. B., Drug Discovery Today 2003, 8, (24), 1128. 9. Alvarez, R.; Velazquez, S.; San-Felix, A.; Aquaro, S.; De Clercq, E.; Perno, C. F.; Karlsson, A.; Balzarini, J.; Camarasa, M. J., J. Med. Chem 1994, 37, (24), 4185. 10. Buckle, D. R.; Rockell, C.; Smith, H.; Spicer, B. A., J. Med. Chem. 1986, 29, (11), 2262. 11. Katritzky, A. R.; Boulto, A. J., Advances in Heterocyclic Chemistry. Academic Press Inc.: New York, 1974; Vol. 16, p 34. Physical 87 12. Genin, M. J.; Allwine, D. A.; Anderson, D. J.; Barbachyn, M. R.; Emmert, D. E.; Garmon, S. A.; Graber, D. R.; Grega, K. C.; Hester, J. B.; Hutchinson, D. K.; Morris, J.; Reischer, R. J.; Ford, C. W.; Zurenko, G. E.; Hamel, J. C.; Schaadt, R. D.; Stapert, D.; Yagi, B. H., J. Med. Chem. 2000, 43, (5), 953. 13. Brockunier, L. L.; Parmee, E. R.; Ok, H. O.; Candelore, M. R.; Cascieri, M. A.; Colwell, L. F.; Deng, L.; Feeney, W. P.; Forrest, M. J.; Hom, G. J.; MacIntyre, D. E.; Tota, L.; Wyvratt, M. J.; Fisher, M. H.; Weber, A. E., Bioorg. Med. Chem. Lett. 2000, 10, (18), 2111. 14. Tullis, J. S.; Van Rens, J. C.; Natchus, M. G.; Clark, M. P.; De, B.; Hsieh, L. C.; Janusz, M. J., Bioorg. & Med. Chem. Let. 2003, 13, (10), 1665. 15. Kaval, N.; Ermolat'ev, D.; Appukkuttan, P.; Dehaen, W.; Kappe, C. O.; Van der Eycken, E., J. Comb. Chem. 2005, 7, (3), 490. 16. March, A., Expected soybean rust invasion leads to first emergency exemption for triazole fungicides in the USA. In Seed Quest http://www.seedquest.com/News/releases/2004/march/8218.htm; Accessed in 03/01/2008 17. Roman, C., MRF Additives. In Management of the Metal Removal Fluid Environment, http://www.aware-services.com/orc/MRF%20additives.htm; Accessed in 03/10/2008. 18. Al-Abdallah, M. M.; Abu-Orabi, S. T., Korrosion (Dresden) 1991, 22, (3), 520. 19. Fox, P. G.; Lewis, G.; Boden, P. J., Corr. Sci. 1979, 19, (7), 457. 20. Gilchrist, T. L., Het. Chem. 3rd ed.; Addison Wesley Longman Limited: Harlow, England, 1997; p 305. 21. Gilchrist, T. L., Het. Chem. 3rd ed.; Addison Wesley Longman Limited: Harlow, England, 1997; p 89. 22. Fleming, I., Frontier Orbitals and Organic Chemical Reactions. Wiley-Interscience: London, 1976. 88 23. Huisgen, R., Angew. Chem. 1955, 67, 439. 24. Huisgen, R., Angew. Chem., Int. Ed. Engl. 1963, 10, 565. 25. Gilchrist, T. L., Het. Chem. 3rd ed.; Addison Wesley Longman Limited: Harlow, England, 1997; p 91. 26. Carey, F. A. S., R. J., Advanced Organic Chemistry Part B: Reactions and Synthesis. . 4th ed.; Kluwer Academic/Plenum Publishers: New York, 2001; p 359. 27. Nantz, M. H.; Zweifel, G. S., Modern Organic Synthesis: An Introduction. W. H. Freeman and Co.: New York, 2007; p 432-433. 28. Kolb, H. C.; Finn, M. G.; Sharpless, B. K., Angew. Chem. Int. Ed. Engl. 2001, 40, 2004. 29. Bemis, G. W.; Murcko, M. A., J. Med. Chem. 1996, 39, 2887. 30. Zinin, N., Ann. 1860, 114, 217. 31. von Pechmann, H., Ber. 1888, 21, 2756. 32. von Pechmann, H.; Wehsarg, K., Ber. 1888, 21, 2992. 33. Baltzer, O.; von Pechmann, H., Just. Lieb. Ann. Chem. 1891, 262, 302-324. 34. Bladin, J. A., Ber. 1893, 26, 545. 35. Dimroth, O.; Fester, G., Ber. Deut. Chem. Ges. 1910, 43, 2219. 36. von Pechmann, H.; Bauer, W., Ber. 1909, 42, 659. 37. Stolle, R., Ber. Deut. Chem. Ges. 1926, 59B, 1742. 89 38. Bladin, J. A., Ber. 1893, 26, 2736. 39. Dimroth, O., Ber. 1902, 35, 1031. 40. Ghigi, E.; Pozzo-Balbi, T., Gazz. Chim. Ital. 1941, 71, 228. 41. Huttel, R., Ber. 1941, (74B), 1680. 42. Dimroth, O., Ann. 1909, 364, 183. 43. Kleinfeller, H.; Bonig, G., J. Prakt. Chem. 1931, 132, 175. 44. Bertho, A., Ber. 1925, 58B, 859. 45. Bertho, A.; Holder, F., J. Prakt. Chem. 1928, 119, 173. 46. Dimroth, O., Ber. 1902, 35, 4046. 47. Dimroth, O., Ann. 1904, 335, 6. 48. Huisgen, R., Angew. Chem., Int. Edit. 1963, 2, 565. 49. Huisgen, R., Angew. Chem., Int. Edit. 1963, 2, 633. 50. Tornoe, C. W.; Christensen, C.; Meldal, M., J. Org. Chem. 2002, 67, (9), 3057. 51. L'Abbe, G., Bull. Soc. Chim. Bel. 1984, 93, (7), 579. 52. Zhang, L.; Chen, X.; Xue, P.; Sun, H. H. Y.; Williams, I. D.; Sharpless, K. B.; Fokin, V. V.; Jia, G., J. Am. Chem. Soc. Comm. 2005, 127, 15998. 53. Sonogashira, K.; Tohda, Y.; Hagihara, N., Tett. Lett. 1975, 16, (50), 4467. 90 54. Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V. V.; Noodleman, L.; Sharpless, K. B.; Fokin, V. V., J. Am. Chem. Soc. 2004, 127, (1). 55. Ahlquist, M.; Fokin, V. V., Organometallics 2007, 26, 4389. 56. L'Abbe, G., Ind. Chim. Bel. 1969, 34, 519. 57. Soghoian, S., Heavy Metal Toxicity, http://emedicine.medscape.com/article/814960overview; Accessed on 04/11/2011. 58. Kingston, H., Microwave-Enhanced Chemistry Fundamentals, Sample Preparation, and Applications. An American Chemical Society Publication Washington, DC, 1997; p 3-53. 59. Kingston, H. M., Introduction to Microwave Sample Preparation. American Chemical Society Washington, D. C., 1988; p 7-32. 60. Hayes, B. L., Microwave Synthesis: Chemistry at the Speed of Light. CEM Publishing: Mathews, NC, 2002; p 12-13. 61. Halter, H. M.; Fiore, J. V.; Cacciola, A. R.; Ito, T. I. Making a reconstituted tobacco product. (Patent No. 5765570). U.S. (1970). 62. Berry, J. G.; Cunningham, F. E., Poultry Science 1970, 49, (5), 1236. 63. Byrne, T. Extraction of Palm Oil. (Patent No. GB 1209675). U.K. (1970). 64. Letourneau, C. U.; Mamrick, W. A.; Sorenson, B. D., Hos. Man. 1964, 98, 35. 65. Wren, J. J.; Nutt, J., J. Sci. Food & Ag 1967, 18, (3), 119. 66. Walradt, J. P.; Lindsay, R. C.; Libbey, L. M., J. Ag. & Food Chem. 1970, 18, (5), 925. 91 67. Gedye, R.; Smith, F.; Westaway, K.; Ali, H.; Baldisera, L.; Laberge, L.; Rousell, J., Tett. Lett. 1986, 27, (3), 279. 68. Giguere, R. J.; Bray, T. L.; Duncan, S. M.; Majetich, G., Tett. Lett. 1986, 27, (41), 4945. 69. Loupy, A., Microwave in Org. Syn. Wiley-VCH: Weinheim, 2002; p 21. 70. Loupy, A., Microwave in Org. Syn. Wiley-VCH: Weinheim, 2002; p 61. 71. Loupy, A.; Petit, J.; Hamelin, F.; Texier-Boullet, P.; Jacquault, D. M., Syn. 1998, (9), 1213. 72. Varma, R. S., Green Chem. 1999, 1, (1), 43. 73. Loupy, A., Microwave in Org. Syn. Wiley-VCH: Weinheim, 2002; p 298. 74. Loupy, A., Microwave in Org. Syn. Wiley-VCH: Weinheim, 2002; p 295. 75. Carruthers, W., Cycloaddition Reactions in Organic Synthesis. Pergamon Press: Oxford, 1990. 76. Xiao, F.; Xu, W.; Qiu, S.; Xu, R., Cat. Let. 1994, 26, 209. 77. Loupy, A., Microwave in Org. Syn. Wiley-VCH: Weinheim, 2002; p 296. 78. Monomode reactors: Prolabo, S. S. a. S. C., STAR system 2 and 6 and Discover. Personal Chemistry, Smith Synthesizer and Smith Creator. 79. Cave, G.; Raston, C. L.; Scott, J. L., Chem. Comm. 2001, 21, 2159. 80. Yadav, L. D. S.; Yadav, B. S.; Dubey, S., Tett. Lett. 2004, 60, (1), 131. 81. Perez, E. R.; Marrero, A. L.; Perez, R.; Autie, M. A., Tett. Lett. 1995, 36, (11), 1779. 92 82. Avalos, M.; Babiano, R.; Bravo, J. L.; Cintas, P.; Jimenez, J. L.; Palacios, J. C., Tett. Lett. 1998, 39, 9301. 83. Pinto, D. C. G. A.; Silva, A. M. S.; Almeida, L. M. P. M.; Carrillo, J. R.; Diaz-Ortiz, A.; de la Hoz, A.; Cavaleiro, J. A. S., Synlett 2003, 10, 1415. 84. Baruah, B.; Prajapati, D.; Boruah, A.; Sandhu, J. S., Syn. Comm. 1997, 27, (15), 2563. 85. Karmakar, D.; Prajapati, D.; Sandhu, J. S., Syn. Comm. 1998, 28, (13), 2415. 86. Garrigues, B.; Laporte, C.; Laurent, R.; Laporterie, A.; Dubac, J., Lieb. Ann. 1996, (5), 739. 87. Loupy, A., Microwave in Org. Syn. Wiley-VCH: Weinheim, 2002; p 317. 88. Loupy, A., Microwave in Org. Syn. Wiley-VCH: Weinheim, 2002; p 323. 89. Loupy, A., Microwave in Org. Syn. Wiley-VCH: Weinheim, 2002; p 333. 90. Marigo, M.; Wabnitz, T. C.; Fielenbach, D.; Jorgensen, K. A., Ange. Chem., Inter. Edit. 2005, 44, (5), 794. 91. Zarudnitskii, E. V.; Ivanov, V. V.; Yurchenko, A. A.; Pinchuk, A. M.; Tolmachev, A. A., Heter. Chem. 2002, 13, (2), 146. 92. Brock, C. P.; Companion, A. L.; Kock, L. D.; Niedenzu, K., Inorg. Chem. 1991, 30, (4), 784. 93. Yoo, H. S.; Lim, J. H.; Kang, J. S.; Koh, E. K.; Hong, C. S., Polyhedron 2007, 26, (15), 4383. 94. Tao, L.; Zhang, L. L.; Shen, S. J.; Han, X. P., Chin. Chem. Let. 2001, 12, (9), 763. 93 95. Appukkuttan, P.; Dehaen, W.; Fokin, V. V.; Eycken v, E., Org. Lett. 2004, 6, (23), 4223. 96. Punna, S.; Diaz, D. D.; Li, C.; Sharpless, K. B.; Fokin, V. V.; Finn, M. G., Polym. Prepr. (Am. Chem. Soc., DiV. Polym. Chem.) 2004, 45, (1), 778. 97. Ermolat'ev, D.; Dehaen, W.; Van der Eycken, E., QSAR & Comb. Sci. 2004, 23, (10), 915. 98. Lermontov, S. A. S., S.V.; and Pushin, A.N., J. Fluorine Chem. 2000, 105, 141. 99. Garanti, L.; Molteni, G., Tett. Lett. 2003, 44, 1133. 100. Gouault, N. C., J. F.; Sauleua, A.; and David, M., Tett. Lett. 2000, 41, 7293. 101. Harju, K.; Vahermo, M.; Mutikainen, I.; Yli-Kauhaluoma, J., J. Comb. Chem. 2003, 5, 826. 102. Carey, F. A.; Sundberg, R. J., Advanced Organic Chemistry Part B: Reactions and Synthesis. . 4th ed.; Kluwer Academic/Plenum Publishers: New York, 2001. 103. Cwiklicki, A.; Rehse, K., Arch. Pharm. Med. Chem. 2004, 337, (3), 156. 104. Lewis, W. G.; Magallon, F. G.; Fokin, V. V.; Finn, M. G., J. Am. Chem. Soc. 2004, 126, (30), 9152. 105. Gonda, Z.; Novak, Z., Dalton Transactions 2010, 39, 726. 106. Molteni, G. a. B., P. D., Tetrahedron 2005, 61, (21), 4983. 107. Laurent, B. A.; Grayson, S. M., J. Am. Chem. Soc. 2006, 128, 4238. 108. Lee, J. K.; Chi, Y. S.; Choi, I. S., Langmuir 2004, 20, (3844). 94 109. Ramachary, D. B.; Barbas, C. F., Chem. Eur. J. 2004, 10, 5323. 110. Khanetskyy, B.; Dallinger, D.; Kappe, C. O., J. Comb. Chem. 2004, 6, 884. 111. Abu-Orabi, S.; Atfah, A.; Jibril, I.; Marii, F.; Ali, A., Gazzetta Chimica Italiana 1991, 121, 397. 112. Ankati, H.; Biehl, E., Tett. Lett. 2009, 50, (32), 4677. 113. Guanti, G.; Rentana, R., Tetrahedron: Asymmetry 2001, 12, 1185. 114. Cheng, H.; Cao, X.; Xian, M.; Fang, L.; Cai, T. B.; Ji, J. J.; Tunac, J. B.; Sun, D.; Wang, P. G., J. Med. Chem. 2004, 48, 645. 115. Zhang, G.; Fang, L.; Zhu, L.; Sun, D.; Wang, P. G., Bioorgan. Med. Chem. 2006, 14, (2), 426. 116. Holzer, W., Tetrahedron 1991, 47, 9783. 117. Abu-Orabi, S.; Atfah, A.; Jibril, I.; Marii, F.; Ali, A., J. Heterocyclic Chem. 1989, 26, 1461. 118. Miller, S. I.; Tanaka, Y., Tetrahedron 1973, 29, 3285. 119. Huisgen, R.; Szeimies, G.; Mobius, L., Chem. Ber. 1967, 100, 2494.