Three-color Western Blot Protocol
advertisement

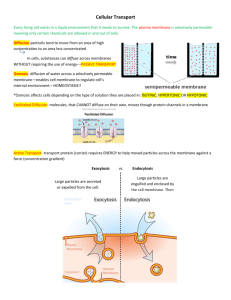
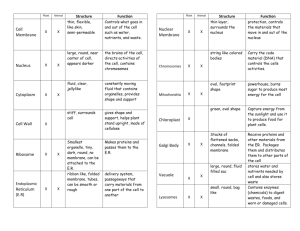
Ari Berman – 12/18/2007 Three-color Western Blot Protocol This protocol assumes that you have a standard western blot protocol in place in your laboratory. The steps provided here are intended to modify that protocol to incorporate three-color fluorescence into your gels. This procedure can also be used with a single or two colors just as easily. I’ve found this system to work much more reliably (and less stressfully) than the standard ECL methods and it removes the need for stripping or reprobing your membranes. Materials needed: Millipore Immobilon-FL PVDF membrane (for fluorescence; Cat: IPFL00010) PBS or TBS Tween-20 Bovine Serum Albumin (BSA) Invitrogen Novex Sharp protein ladder (shows up under fluorescence) Three Primary antibodies – Rabbit, Mouse, Sheep hosts (or other combination) Donkey Anti-rabbit IgG – AlexaFluor 488, Donkey Anti-mouse IgG – AlexaFlour 555, Donkey Anti-sheep IgG – AlexaFlour 633 (or appropriate combination for primaries, the key is 488, 555, and 633 excitation wavelengths. See Figure 1 at the end of the protocol for the spectra analysis) Procedure: * - Note: all volumes listed are for a single membrane. Scale up appropriately for more membranes. 1. 2. 3. 4. 5. 6. 7. Collect cell lysate, load and run gel as specified in laboratory protocol. Cut appropriate sized piece of Immobilon-FL membrane for gel. Place membrane in methanol for 10 seconds to activate, avoid bubbles. Equilibrate membrane in transfer buffer for at least 10 minutes. Equilibrate completed gel in transfer buffer for 5 minutes. Transfer according to lab protocol. Remove transferred membrane from transfer apparatus and place immediately in PBS. 8. Rinse 2x quickly (manual agitation, ~15 sec) with PBS to remove methanol from membrane. 9. Make blocking buffer as follows: 10mL/membrane 1xPBS, 0.1% Tween-20, 5% BSA (w/v) o Pre-mix PBS and tween until in solution o Add powdered BSA to pre-measured volume of PBS-Tween o Vortex at full speed until last traces of BSA are no longer visible (solution should be yellowish, but clear) o Push blocking buffer through syringe filter (0.20um) into clean tube o Use mixture for duration of experiment, store at 4°C when not in use Ari Berman – 12/18/2007 10. Add 5mL blocking buffer to membrane and block at room temperature rocking at 40rpm for 1 hour. 11. Mix primary antibodies with 2mL of blocking buffer for incubation. o Make sure that each of your primary antibodies are from different hosts and that you have secondary antibodies to match those hosts. With proper blocking, the same host as the host tissue may be used without much trouble. o Be sure to vortex antibody mixture thoroughly. 12. Place blocked membrane in heat-sealed polypropylene pouch. 13. Cut one corner from the pouch and add the 2mL of antibody mixture to the pouch. 14. Remove any large bubbles from the pouch and re-seal cut corner. 15. Incubate overnight at 4°C rocking at 40rpm. 16. Cut a corner from the pouch and drain antibody mixture into a tube for storage, mix with 0.05% sodium azide. Can reuse antibody mixture 3x. 17. Cut the membrane out of the pouch and place in 4-6mL 1x PBS, 0.1% Tween-20 (wash buffer). 18. Manually agitate the membrane, then lift it out of the wash buffer with forceps and drain the liquid from the washing container. 19. Replace the membrane and wash 3x10min in wash buffer rocking at room temperature. 20. Remove blocking buffer from 4°C storage and equilibrate to room temperature. 21. During the final wash, mix 4mL blocking buffer and appropriate secondary antibodies at 1:1000 dilution (must be AlexaFluor 488, 555, and 633 to work well with Typhoon). 22. Vortex thoroughly, store in the dark until use (wrap tube in foil). 23. Drain the final wash from the membrane container. 24. Add the 4mL of secondary antibody mixture to the membrane and wrap the container in aluminum foil in a light-proof manner to avoid photobleaching of fluorescent dyes. 25. Incubate membrane at room temperature for 2 hours rocking. 26. Wash the membrane as in steps 22-23, re-wrapping the container in foil during each wash (it is vital to minimize the membranes exposure to light once the fluorophores are in contact with the membrane through the end of the experiment). 27. After the final wash, rinse briefly in PBS and place membrane on a piece of Whatman paper protein side up, in a drawer (out of the light) to dry for 1 hour. 28. Wrap the membrane carefully in foil and take the dried membrane to the Typhoon 9810 in Bldg 1, rm 117. o Note: You must be trained and checked out by Christina Ferrell in order to use the Typhoon. 29. Open the typhoon control system and allow the lasers to warm up. 30. Wipe down the platen of the Typhoon with the dust cloth located on top of the machine. 31. Place the dried membrane protein side down onto the platen with the top (wells) facing the left side of the platen. 32. Close the lid and select the proper area to be scanned 33. Select Fluorescence as the acquisition mode, then click Setup. 34. Select the 520 BP 40 emission filter from the drop-down list and change the PMT to 300. Make sure the selected laser is Blue2 (488). Ari Berman – 12/18/2007 35. Click the second checkbox and select 580 BP 30, set PMT to 300. Make sure the 532nm laser is selected. 36. Click the third checkbox and select 670 BP 40, set PMT to 300. Make sure the 633nm laser is selected. 37. Click OK. 38. Select the third “R” from the left on the bottom row from the orientation menu. 39. Set the Typhoon to “press sample” and to scan at 200um. 40. Select “FlourSep” from the image analysis menu below the user comment field. 41. Once the typhoon reads ready, click SCAN. o The Typhoon will scan your blot three times, once at each wavelength 42. When the scanning is complete, your image will be transferred to the fluorsep program. 43. Right-click on your image and select “Actual Size” to zoom out. 44. Change the view to “side-by-side” view (seventh button from left on top toolbar). 45. Click on “F1” box and resize it to fit the average band size on your gel. All boxes will match the size change. 46. Move the F1 box to one of the bands in channel 1. 47. Do the same with the F2 box in channel 2, and the same for F3 in channel 3. 48. Click “Perform Separation”. 49. Exit Fluorsep and load up ImageQuant. 50. Select your file from inside the correctly named folder that the images were saved under from the scanning program. 51. Right-click on the image and select “Gray/Color adjust”, adjust the image to minimize background and brighten the bands of interest. Do this for each channel. 52. Click OK. You should see your finalized gel with three colors in front of you. 53. You can either quantify using ImageQuant, or export the images for use with other programs. 54. To export the color image, go to Edit->Copy Image. o Load Microsoft Powerpoint. o Paste the image into powerpoint. o Either save as a powerpoint file, or export it as a tif file. 55. To export the individual channels for quantification, change to side-by-side view as was done in FluorSep. 56. Click once on the channel 1 box, then go to File->Save As and select tif from the “Save as type” dropdown menu. Repeat the procedure for each of the channels. 57. Save the images to a USB drive or to the shared network drive if you have access to that system 58. Quantify as you would a normal gel. Ari Berman – 12/18/2007 Figure 1 – Spectra analysis with Typhoon lasers and filters for AlexaFluors called for in this protocol Figure 2 – An example of a successfully completed three-color western blot Please email Ari Berman at ari@bermanism.com with any questions about this protocol.