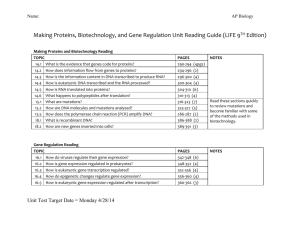
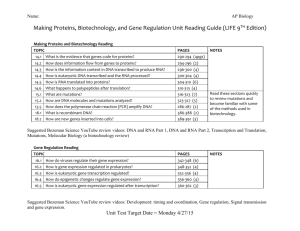
INTRODUCTION
advertisement

Chap 5 Manipulation of Gene Expression in Procaryotes I. Introduction A major objective of gene cloning is the expression of the cloned gene to study the biologic functions or to produce recombinant proteins (i.e. insulin). But gene cloning doesn’t guarantee successful expression. Factors that influence gene expression 1. The nature of the transcriptional promoter and terminator sequences 2. The strength of ribosome binding site 3. Efficiency of translation (mRNA stability, mRNA secondary structure..) 4. # of the cloned gene copies (or # of plasmids) and whether the gene is plasmid borne or integrated into the chromosome. 5. Nature and cellular location of the expressed protein (intra- or extracellular? secreted? toxic?) 6. Post-translational processing: glycosylation, proteolytic processing… 7. The intrinsic stability of the protein (misfolding of the proteins? susceptible to proteolysis?) A large fraction of proteins (varying from 30% to 70% of all proteins made) is immediately degraded after synthesis before forming functional proteins8. These socalled DRiPs (defective ribosomal products) are the result of defective transcription or translation, alternative reading frame usage, failed assembly into larger protein complexes, the incorporation of wrong amino acids owing to mistakes by aminoacyl-tRNA synthetases or altered ubiquitin modifications. DRiPs are immediately degraded to prevent the formation of protein aggregates, which would affect cell viability. Ubiquitination is a post-translational modification in which ubiquitin, a 76–amino acid protein, is covalently added to lysine residues. In humans, the ubiquitination reaction is catalyzed by >500 E3 ligases, each of which transfers ubiquitin 1 to specific protein targets. There are several types of ubiquitin modification, and these may have different effects on target proteins. The best known is the polyubiquitin chain, which targets proteins for proteasomal degradation. The polyubiquitin chain begins with a ubiquitin conjugated at its C terminus to a lysine residue in a target protein. 26S proteasome: A giant multicatalytic protease that resides in the cytosol and the nucleus. The 20S core, which contains three distinct catalytic subunits, can be appended at either end by a 19S cap or an 11S cap. The binding of two 19S caps to the 20S core forms the 26S proteasome, which degrades polyubiquitylated proteins into peptides1. Some of the above factors can be improved by proper design (i.e. select strong promoter, use multiple gene copies…) Choice of expression system is very important!!! (May affect location of proteins, poor translational processing, poor transcription, protease-mediated degradation) Major expression systems are classified into procaryotic and eucaryotic. Procaryotic (e.g. E. coli): Pros: 1. Very well-studied and common in protein production. 2. Grow fast (doubling time20 min), grow easily easy fermenter operation. 3. Normally high yield (high cell density). 4. Minimum media (simple composition, e.g. Na+, K+, Mg2+, Ca2+, NH4+, Cl-, SO42-, glucose and carbon sources) cheap. Cons: 1. Often fail to perform suitable post-translational modifications. 2. Inclusion body (insoluble proteins) when overexpressing makes purification and regaining of protein conformation (protein renaturation) more difficult. Eucaryotic: next chapter II. Strong and regulatable promoters 1 Why strong promoters? Some endogenously expressed proteins (e.g. viral proteins or tumor proteins) are degraded to short peptides and routed to MHC class I in ER, where the formed complex leaves ER to the surface for presentation. 2 Has higher affinity for RNA pol so the downstream gene is highly (frequently) transcribed. Why regulatable promoters? Continuous overexpression of a cloned gene is often detrimental to the host cell because it drains the energy and other resources and impair cellular functions. genes are constructed under strong and regulatable promoters. genes are expressed only when “induced”. Examples 1. E. coli lac promoter: (a) Regulated by IPTG Cells are grown in the absence of lactose and repressor binds to the operatorgenes can’t be transcribed. Only when IPTG is added then starts the gene expression. Lac repressor promoter operator Gene 1, 2,3,.. Induction (turn on) by IPTG (isopropyl--D-thiogalactopyranoside) IPTG prevents lac repressor from binding to the operator Transcription occurs Very common (b) regulated by CAP (catabolite activator protein) 3 cAMP CAP promoter operator Gene 1, 2,3,.. Binding of cAMP to CAP further enhances the affinity for RNA pol *level of cyclic AMP is highest when glucose level is low cAMP CAP promoter operator Gene 1, 2,3,.. RNA pol Combining the above, induce protein expression at high IPTG (or lactose) and low [glucose]. (high [cAMP])highest transcription. 2. Trp promoter: (regulates the transcription of genes responsible for Trp synthesis) off (negatively regulated): tryptophan-trp repressor protein complex binding to trp operator transcription shutdown 3. on (positively regulated): removal of tryptophan Bacteriophage T7 promoter: T7 RNA pol IPTG (to T7 promoter Target gene lac promoter T7 RNA pol gene induc T7 promoter is very strong, but requires T7 RNA pol to activate. e Two recombinant genes can be co-introduced into the cells for expression. Alternatively, the genes encoding T7 RNA pol can be integrated into the chromosomal DNA to form a stable cell line. 4. pL promoter (from bacteriophage ): 4 Controlled by cI repressor protein Cells carrying temp-sensitive cI repressor are grown at 28C (cI repressor is expressed under its own promoter pCI at 28C) cI repressor prevents transcription when CD is high enoughincrease to 42C thermosensitive cI repressor is inactivated transcription is on. Effectiveness of deactivating a repressor depends on # of repressor # of copies of promoter sequences ratio too large difficult to induce ratio too small transcription is “leaky” (transcription occurs in the absence of inducer) Strategy: Put repressor genes in a plasmid: low copy # (e.g. 1-8 copies/cell) Put promoter-target gene in another plasmid: high copy number (e.g. 30-300 copies/cell) maintain the ratio to effectively deactivate and activate. III.Expression vectors Regulatable, strong promoters may not guarantee high yield of gene products. Efficiency of translation, stability of protein, etc. also are factors2. Expression vectors are similar to cloning vectors but contain more elements to confer efficient expression. e.g. The expression plasmid pKK233-2 contains: tac promoter (a hybrid that includes the -10 region of lac promoter and -35 region of trp promoter, can be induced by IPTG, 3X and 10X stronger than trp and lac promoters, respectively) RBS, ori. (RBS: a sequence of 6-8 nt (e.g. UAAGGAGG) in mRNA that can base pair with rRNA on the ribosome, generally, binding of mRNA to rRNA increases, the translation initiation increases) 2 Not all mRNA are translated in the same efficiency, differential translation and transcriptional regulation enable the cells to adapt to different stresses (environmental, heat shock, oxygen…) 5 An ATG start codon about 8 nt downstream from the RBS (optional) Multiple cloning site Ampr gene as a selectable marker Note: the RNA sequence from RBS to the first few codons of the cloned gene must not form intrastrand loops, which hampers the binding to ribosome DNA sequence is written as the coding strand, so ATG is often seen as the starting point. IV. Fusion Proteins Problems: yield of foreign proteins normally low for various reasons (e.g. degradation by proteases) Solution: covalently attach the cloned gene product to a stable (host) protein to form a fusion protein to protect the desired recombinant protein. Construct at DNA level transcribed RNA must have correct base sequence (stop codon in the middle must be eliminated) Reading frame must be correct, base sequence in the linker must be precise, otherwise ORF will be wrong (need to know the precise sequence of these two proteins) Cleavage of fusion proteins The fusion may not be suitable as the final product because: The biological function might be lost 6 Stringent regulation by government agencies (e.g. FDA) The EK cleavage site enables the cleavage of the fusion by enterokinase at the specified site. Another linker often used is the Xa linker (Ile-Glu-Gly-Arg) which can be recognized by a blood coagulation factor (Xa) and specifically recognized at the C-terminus the desired protein should therefore be in the second segment. Applications of fusion proteins (many applications, give one example only) e.g. simplify purification dual function of the fusion: reduce the degradation, enable the cleavage Flag (a peptide recognized by EK) -Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys-IL2 IL2: a cytokine that stimulates both T-cell growth and B-cell Ab synthesis enable the product to be purified by immunoaffinity chromatography in which MAb directed against Flag is immobilized on a polypropylene support and used as a ligand to bind the fusion. Unidirectional tandem gene arrays Generally, gene expression increases as plasmid copy # increases, but other plasmidencoding proteins (e.g. antibiotics resistance) are over-expressed too wasting metabolic energy, constraining cellular activities increasing copy # is not always effective tandem gene arrays (cloning multiple copies of genes into a low copy # plasmid) problem: simple end-to-end ligation of DNA results in a random orientation of the arrays of genes 7 The gene, together with the translational start and stop signals, is cloned into the EcoRI site. The gene is cut by Ava I the DNA fragments have non-identical sticky endsligation results in unidirectional array. V. Golden Gate Shuffling: A One-Pot DNA shuffling Method Limitations of the traditional cloning methods 1. Time consuming 2. Inefficient 3. Require the unique restriction sites become limited for large recombinant DNA molecules. Golden Gate Shuffling is a protocol to assemble separate DNA fragments together into an acceptor vector in one step and one tube. The principle of the cloning strategy is based on the ability of type IIs restriction enzymes to cut outside of their recognition site. 1. Two DNA ends terminated by the same 4 nucleotides. (sequence f, composed of nucleotides 1234, complementary nucleotides noted in italics) 2. Sequence f are flanked by a BsaI (type II restriction enzymes) recognition sequence, B. 3. The type IIs restriction enzymes removes the enzyme recognition sites and generates ends with complementary 4 nt overhangs. 4. These ends can be ligated seamlessly, creating a junction that lacks the original site. 8 One-pot one-step assembly of a GFP construct from 10 constructs. 1. A DNA shuffling protocol would consist of first selecting a number of 4 nucleotides ‘recombination sites’ on a nucleotide sequence alignment of several homologous genes. 2. The selection of these recombination sites defines modules that consist of a core sequence (C1-C9) flanked by two 4 nt sequences. 3. These modules can be amplified by PCR with primers designed to add flanking BsaI sites on each side of the modules (the BsaI cleavage sites perfectly overlapping with the recombination sites) 4. The recipient expression vector, pX-LacZ contains two BsaI sites compatible with the first (C1) and last (C9) modules. 5. Incubation for 2 minutes at 37℃ and 5 minutes at 16℃, both steps repeated 50 time. 6. Incubation for 5 minutes at 50℃ (final digestion). 7. Incubation for 5 minutes at 80℃ (heat inactivation). Increasing protein stability Normally, the half lives of proteins range from a few minutes to hours (some exceptions exist, e.g. collagen has an half life of years), because cells contain proteases that degrade unnecessary and 9 abnormal proteins. This enables the cells to recycle the resources, a housekeeping function to keep the cell viability. Normally, proteins with more disulfide bonds (S-S between Cys) and certain amino acids at the N-terminus are more stable more proteins accumulate and the yield increases. Ex: stability of -galactosidase with certain a.a added to the N-terminus Strategies: Change the a.a. at the N-terminus Increase the number of S-S bonds Co-express chaperone proteins (e.g. groEL, dna J, dna a.a. added Half life Met, Ser, Ala > 20 h Thr, Val, Gly > 20 h Ile, Glu > 30 min Arg 2 min K….) to aid the protein folding VI. Overcoming O2 Limitation Oxygen is generally required for cell growth, to support respiration and maintain cellular functions and protein expression, but oxygen’s solubility is low. If the CD is high, even larger amount of air or oxygen or increasing the stirring speed may not be enough. When O2 depletion occurs, cells would enter stationary phase and die eventually. If engineering approaches fail, what can we do? bacterial hemoglobin Solve: bacterium “Vitreoscilla” lives in stagnant ponds (oxygen deficient). To obtain oxygen for growth and metabolism, the bacteria express a hemoglobin-like protein that fetch oxygen from the environment and transport into the cells. When this gene is cloned and expressed in E. coli, the recombinant E. coli shows higher metabolic activity and higher protein production at low levels of O2. VII. DNA Integration into the Host Chromosome Why integrate DNA into the chromosome? Plasmid-borne expression drains the cellular energy due to the antibiotics-resistant and other genes are expressed, plasmid replication requires the resources and energies too. Plasmid instability: plasmid-free cells outgrow plasmid-bearing cells, so after several passages, the percentage of cells bearing plasmids drops, protein production drops as well. If the foreign DNA is on the chromosome, it is not as easy to occur. 10 How? 1. Choose a suitable integration site. 2. Clone part of the chromosomal DNA sequence at the integration site into the vector (e.g. plasmid). The chromosomal DNA sequence on the vector and at the integration site must be similar in sequence, at least 50 bp, so that homologous recombination can occur. 3. Clone the target gene (and promoter) into the plasmid (vector) flanked by the chromosomal DNA sequence. 4. Transfer the plasmid into a host cell (The vector does not replicate within the host cell). 5. Select the host cells that have the target gene integrated into the chromosome. More details discussed in the eucaryotic cell Increasing Secretion Secretion is important for many human proteins (e.g. adrenaline, growth factors and many other blood proteins are secreted). In industry, it’s often desired that the proteins be secreted because: The stability of a cloned gene in E. coli depends on its cellular location, stable. secreted proteins tends to be more For example, a recombinant proinsulin is 10X more stable if exported into the periplasm (the space between the inner and the outer membrane). Secreted proteins may give higher purification recovery yield because they are free from thousands of cellular proteins. Drawback: recombinant protein concentration in the medium is low. 11 Secreted proteins have a signal peptide at the Nterminus, facilitating the protein transport though the secretory pathway. When crossing the membrane, the signal peptide is cleaved by peptidase to become the mature protein. Problem: E. coli and other Gram negative bacteria have outer membranes, which prevent proteins from secreting to the medium. Solve: Use gram-positive or eucaryotes which do not have outer membranes (but Gram-negative bacteria such as E. coli is usually excellent first-choice). Fuse a signal peptide or engineer a fusion protein with signal peptide at the N-terminus Lower the expression level because sometimes over-expression could overload the secretion machinery, thus mitigating the secretion. Co-express a limiting factor in the secretion pathway. For instance, clone prl A4 and secE genes (which encode the major components of the molecular apparatus that moves proteins across the membrane) into E. coli % of secreted (and mature) protein increases from 50% to 90%. Clone bacteriocin release protein: bacteriocin (in Gram negative bacteria) is secreted with the help of bacteriocin release protein, which permeabilize the inner and outer membranes Co-express this protein with the target protein under the control of the same promoter. 12 IX. Reducing the Metabolic Load Expression of foreign gene often changes the metabolism and impairs the normal cellular function, due to the increased metabolic load (burden) for the following reasons: Competing for amino acids, tRNA and energy (ATP). DO is often insufficient for cell metabolism and plasmid maintenance. Increasing plasmid copy number often requires increasing amounts of cellular energy for plasmid replication and maintenance. Foreign proteins may jam the export sites and impair proper localization of host proteins. Foreign proteins may be toxic to the cells. Outcomes: Plasmid instability: cells w/o plasmid outgrow cells with plasmid loss of recombinant plasmid. Energy intensive processes such as nitrogen fixation and protein synthesis slow down. Translational error: because tRNA could be limiting so incorrect a.a. may be incorporated (chance could be 10 times more when overexpression occurs). Solution: Use low copy # plasmid instead of high copy number plasmid. Integrate the foreign DNA into the chromosome. Use strong, regulatable promoter, so cell culture is divided into two phases: Growth phase: cell growth without target protein expression Production phase: when CD is high, production is induced (e.g. by IPTG, heat…) Express at a modest level (e.g. 5% of the total protein), but at high CD in fermentation. High expression and high CD are not easy to achieve simultaneously. Expression at high level (15%) but at low CD may not be more efficient. X. Appendix Inclusion body (IB) 13 Protein aggregates that usually lack biologic functions separate the IB by centrifugation or filtration (may facilitate purification)IB denaturation then renaturation. Denaturation Performed by strong acid, strong base, high temperature and pressure, proteases etc. Usually chemical agents are used: Urea: 8-10 M, destroy H-bond and hydrophobic interaction. Guanidine hydrochloride (GuHCl): 6-8 M, disrupts hydrophobic and ionic interactions. Dithiothreitol (DTT), -mercaptomethanol: disrupt the S-S bond. EDTA or EGTA: chelate metal ions to avoid unwanted chemical reactions. Renaturation (refolding) Dialysis: change the buffer and dilute the denaturant concentration gradually. Renaturation buffer: usually contains Tris-HCl (pH buffer), low concentration of denaturant (e.g. urea) to prevent aggregation and oxidizing agent to oxidize the –SH group for S-S bond formation. 14