Supplementary Information 2 (doc 41K)
advertisement
")
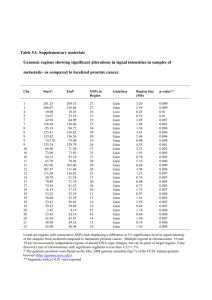
Bioinformatics analysis Methods Bioinformatics analysis was performed to assess the biological relevance of significant individual SNPs. For non-coding SNPs, evolutionary conservation in 17 vertebrates was examined using the “Vertebrate Multiz Alignment1 & Conservation” track on the UCSC Genome Browser.2 The conservation track is based on a phylogenetic hidden Markov model, phastCons.3 We determined whether the associated SNP fell within a predicted evolutionarily conserved element using the phastCons conserved elements track.4 Finally, we used the MATCHTM program5 (http://www.gene-regulation.com/cgi-bin/pub/programs/match/bin/match.cgi) to determine whether non-coding SNPs located in putative regulatory regions create or destroy any TRANSFAC®6 predicted transcription factor binding sites. Two sequences (101bp each), containing the alternate alleles for the SNP of interest, were submitted for analysis. In addition to the default settings, MATCH was set to search only the vertebrate matrix groups and to minimize the false positive matches. Results The three signficant SNPs (56, 126 and 131) were subjected to bioinformatics analysis to assess their biological significance. Neither SNP 56 nor SNP 126 is conserved across species, as determined by examination of the Vertebrate Multiz Alignments and Conservation track on UCSC (http://genome.ucsc.edu/cgibin/hgGateway). SNP 131 does fall within a region of appreciable conservation, but it does not correspond to any phastCons-predicted conserved elements. For the vertebrate sequences with which it could be aligned (human, chimp, rhesus, dog, cow and elephant), the risk allele of SNP 131 (T) is conserved, suggesting that this is unlikely to be the deleterious allele. Finally, none of the three SNPs appeared to create or abolish a TRANSFAC predicted transcription factor binding site, as determined by the MATCHTM program.5 References 1. Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 2004 Apr; 14(4): 708-715. 2. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM et al. The human genome browser at UCSC. Genome Res 2002 Jun; 12(6): 996-1006. 3. Siepel A, Haussler D. Combining phylogenetic and hidden Markov models in biosequence analysis. J Comput Biol 2004; 11(2-3): 413-428. 4. Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 2005 Aug; 15(8): 1034-1050. 5. Kel AE, Gossling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, Wingender E. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res 2003 Jul 1; 31(13): 3576-3579. 6. Wingender E, Chen X, Fricke E, Geffers R, Hehl R, Liebich I et al. The TRANSFAC system on gene expression regulation. Nucleic Acids Res 2001 Jan 1; 29(1): 281-283.