5-BRADFORD PROTEIN ASSAY
advertisement

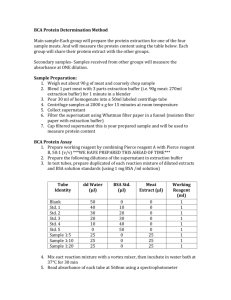
BRADFORD PROTEIN ASSAY Background & Theory Four spectroscopic methods are routinely used to determine the concentration of protein in a solution. These include measurement of the protein's intrinsic UV absorbance and three methods which generate a protein-dependent color change; the Lowry assay, the Smith copper/bicinchoninic assay and the Bradford dye assay. The first, UV absorbance, requires that a pure protein with known extinction coefficient be used, in a solution free of interfering (UV absorbing) substances. The Lowry and copper/bicinchoninic assays are based on reduction of Cu2+ to Cu1+ by amides. Although this makes them potentially quite accurate, they require the preparation of several reagent solutions, which must be carefully measured and mixed during the assay. This is followed by lengthy, precisely timed incubations at closely controlled, elevated temperatures, and then immediate absorbance measurements of the unstable solutions. Both assays may be affected by other substances frequently present in biochemical solutions, including detergents, lipids, buffers and reducing agents. This requires that the assays also include a series of standard solutions, each with a different, known concentration of protein, but otherwise having the same composition as the sample solutions. The Bradford assay is faster, involves fewer mixing steps, does not require heating, and gives a more stable colorimetric response than the assays described above. Like the other assays, however, its response is prone to influence from non protein sources, particularly detergents, and becomes progressively more nonlinear at the high end of its useful protein concentration range. The response is also protein dependent, and varies with the composition of the protein. These limitations make protein standard solutions necessary. Objective This assay is based on the use of a dye, Coomassie Brilliant Blue G-250, to which protein binds, altering the light absorbance properties of the dye. When the dye is prepared as an acidic solution (in 85% phosphoric acid), it maximally absorbs light with a wavelength of 465 nm. Addition of protein results in a shift of the dye's absorption maximum to 595 nm. As the protein concentration increases, the absorbance of light at 595 nm increases linearly. This increase in absorbance can be measured in a spectrophotometer. Although the absorbance of Coomassie blue dye at 595 nm is proportional to the amount of protein bound, it is necessary to establish a correspondence between absorbance values and known amounts of protein. To do this, you will prepare a series of protein standards – dilutions of a protein solution of known concentration. Once you have measured the A595 of each standard, you will be able to plot the A595 as a function of the known protein content of each standard. After measuring the A595 of unknown sample, the standard curve then can be used to determine the amount of protein corresponding to the absorbance values measured. Protocol Preparation of bovine serum albumin protein assay standards: In order to measure and plot a standard curve of protein concentration versus absorbance at 595 nm, a series of dilutions of the BSA protein standard stock solution must be prepared. The easiest way to solve for the volume of protein stock solution required for each dilution is to use the formula C1V1 = C2V2. C1 is the concentration of the protein stock solution, V1 is the volume of the stock solution required, C2 is the concentration of the diluted sample, and V2 is the volume of the diluted sample. The concentration of the stock solution (C1) is 100 µg/ml, the concentration of the diluted sample is (C2), and the volume of the diluted sample is fixed at 200 µl. Therefore, solving for the volume of stock solution required: V1 = C2V2/C1 Protein Standards - Protein standards should be prepared in the same buffer as the samples to be assayed. A convenient standard curve can be made using bovine serum albumin with concentrations of 0, 10, 20, 30, 40, 50 µg/ml for the microassay (extinction coefficient of BSA is 0.667). Bradford Reagent - Bradford reagent can be made by dissolving 100 mg Coomassie Blue G-250 in 50 ml 95% ethanol, adding 100 ml 85% (w/v) phosphoric acid to this solution and diluting the mixture to 1 liter with water. Procedure 1. Prepare a 10-fold dilution of a 1 mg/ml BSA sample by adding 100 µl of 1 mg/ml BSA to 900 µl of distilled water to make 100µg/ml BSA. 2. Generate test samples for the reference cell, blank, BSA standards and the protein sample to be tested according to Table 1 in disposable cuvettes. 3. Note that a dilution of the protein sample may be required for the resulting absorbance to fall within the linear range of the assay. 4. Allow each sample to incubate at room temperature for 5 minutes. 5. Measure the absorbance of each sample at 595 nm using a UV-visible spectrophotometer. Be sure to allow the instrument to warm up for at least 15 minutes prior to use. 6. Plot the absorbance of each BSA standard as a function of its theoretical concentration. The plot should be linear. Determine the best fit of the data to a straight line in the form of the equation "y = mx + b" where y = absorbance at 595 nm and x = protein concentration. 7. Use this equation to calculate the concentration of the protein sample based on the measured absorbance. If the absorbance of the test sample is outside of the absorbance range for the standards, then the assay must be repeated with a more appropriate dilution, if any. The linear range for the assay (and for most spectrophotometers is 0.2 - 0.8 O.D. units). Table 1: Preparation of test samples for the Bradford protein assay. Test Sample Blank BSA Standard (10 µg/ml) BSA Standard (20 µg/ml) BSA Standard (30 µg/ml) BSA Standard (40 µg/ml) BSA Standard (50 µg/ml) Protein Sample Sample vol., Water, µl µl 0 200 20 180 Bradford reagent, µl 800 800 40 160 800 60 140 800 80 120 800 100 100 800 100 100 800