Cell and Molecular Biology
Summer semester practice, part I
1. FLUORESCENT STAINING OF F-ACTIN, G-ACTIN AND
DNA IN CELLS
1)
Fix the cells growing on the cover slip with formaldehyde solution - pour formaldehyde
solution to a Petri dish with a cover slip and incubate for 10-15 minutes.
Formaldehyde crosslinks proteins in cells and fixes them in the same positions they had
in living cells. To avoid cell lysis during sample preparation it is necessary to work with
solutions that are isoosmotic. This is the reason why formaldehyde is diluted in PBS
(phosphate buffered saline). Formaldehyde solution contains also Triton X-100, a
detergent that causes permeabilization of cell membranes and thus enables access of
substances used for staining into cells.
2)
Pour out the formaldehyde solution from the Petri dish to the waste container.
3)
Wash the cover slip with cells (at least) 3 x 5 minutes in PBS. “Washing” means: pour
approximately 5 ml of PBS into the Petri dish, incubate approximately 5 minutes and
pour out the PBS from the Petri dish to the waste container. Repeat this procedure at
least three times.
During this washing step the rests of formaldehyde are diluted out from the sample by
repeated washing with PBS. Formaldehyde when present in sample produces unspecific
background fluorescence. It would also cause crosslinking of proteins used in next steps
of the staining protocol (e.g. antibodies) leading to unspecific staining.
4)
Pour out PBS and dry surroundings of the cover slip in the dish. Then put a drop of
staining solution containing phalloidin and DNase I (approximatelly 100 l) on the cells
on the cover slip. Cover the drop with a square of parafilm, incubate 20 minutes.
Phalloidin is a poison from mushroom Amanita phalloides that specifically binds to Factin. This is also the purpose of its toxicity at the cellular level - it stabilizes actin
filaments and disturbs cellular processes that are dependent on actin dynamics. For
fluorescent detection of actin filaments, we use in our protocol phalloidin that is
covalently linked to an excitable fluorescent molecule (=fluorophor) called TRITC
(tetramethyl rhodamine isothiocyanate) which emits red light after excitation.
Microfilaments stained with phallodin-TRITC conjugate will be therefore seen in
microscope in red. DNase I binds to G-actin in cells. We use in our protocol DNase I
that is conjugated with Alexa Fluor 488 and emits green light after excitation. Free
actin molecules stained with DNase I-Alexa Fluor 488 will be therefore seen in green.
Covering the staining solution with a square of parafilm minimizes evaporation and
helps to spread the staining solution over the whole cover slip.
5)
Wash the cover slip with cells (at least) 3 x 5 minutes in PBS.
During washing the cells get rid of unbound phalloidin and DNase I that would cause
unspecific background fluorescence when present in the sample.
6)
Transfer the cover slip on a slide with a drop of mounting medium, cells facing down.
Mounting medium contains DAPI (4',6-diamidino-2-phenylindole) that binds to dsDNA
(double stranded DNA). After binding to DNA it is excitable by UV light and emits light
in blue.
Cell and Molecular Biology
Summer semester practice, part I
2. COMPARISION OF ACTIN AND MYOSIN EXPRESSION IN
DIFFERENT TYPES OF TISSUES
2.1. ISOLATION OF PROTEINS FROM DIFFERENT TISSUES
1)
Sign three 1.5-ml tube: write the number of your study group, the type of the tissue and
some identification of your working group within the study group.
2)
Pipette 700 l of lysis buffer to each of these tubes.
3)
With a scalpel cut several small pieces of the tissue - as small pieces as possible.
Desintegration of the tissue to small pieces is necessary for good contact of the tissue
with lysis buffer and effective lysis.
4)
Transfer the desintegrated tissue to the tubes with the lysis buffer.
Lysis buffer contains SDS (sodium dodecylsulfhate) - detergent that lyses cell
membranes structures, and other components that set up the appropriate pH.
5)
Vortex the content of the tubes.
Vortexing facilitates desintegration of the tissue and makes the lysis more effective.
6)
Incubate the samples for 30-60 minutes on ice, vortex the tubes several times during the
incubation.
Cell proteases released from the tissue during lysis are inhibited when samples are
incubated on ice.
7)
Centrifuge the tissue samples at 14 000 rpm for 5 minutes at 4°C.
After centrifugation, supernatant (=lysate) contains proteins released from lysed cells
and sediment (=pellet) contains unlysed tissue and the rests of cells, i.e. fragments of
cell membranes, organels, nuclei,…
8)
Sign three new 1.5-ml tubes: write the number of your study group, the type of the
tissue and some identification of your working group within the study group.
9)
Transfer carefully approximatelly 200 l of the lysate (=supernatant) to these tubes,
avoid transferring of any pieces of the unlysed tissue from the sediment.
10) For each tissue sample prepare 50 l of 10x diluted lysate to a 0.5 ml tube: mix 45 l of
distilled water with 5 l of lysate, mix well with vortexing.
These samples will be used for determination of protein concentration by Bradford
method.
11) Freeze the residuum of the lysate at -20°C.
Samples can be stored at -20°C for several weeks.
Cell and Molecular Biology
Summer semester practice, part I
2.2. DETERMINATION OF PROTEIN CONCENTRATION BY
THE BRADFORD ASSAY
PRINCIPLE:
The Bradford Reagent can be used to determine the concentration of proteins in
solution. The procedure is based on the formation of a complex between the dye, Brilliant
Blue G, and proteins in solution. The protein-dye complex causes a shift in the absorption
maximum of the dye from 465 to 595 nm. The amount of absorption is proportional to the
protein content. The Coomassie blue dye binds to basic and aromatic amino acid residues,
especially arginine.
For determination of protein concentration in unknown samples by this method it is
necessary to prepare a serial dilution of a protein solution with a known concentration. These
samples with known protein concentration are mixed with Bradford reagent. After measuring
of absorbance of these serial dilutions, the calibration curve for the dependence between
absorbance and concentration of protein can be created. Then concentration of proteins in
unknown samples can be easily calculated from the equation of the calibration curve in the
case we know the absorbance of these samples. The dependence between absorbance and
concentration is linear and is based on the Lambert-Beer´s principle. As a protein standard for
construction of calibration curve bovine serum albumine (BSA) is usually used.
1)
Sign tubes with 1ml of Bradford reagent with type of the tissue.
2)
Pipette 10 l of 10x diluted protein samples into corresponding tube with Bradford
reagent. Vortex the tubes well
The Bradford solution turns blue if proteins were present in the tissue sample.
3)
Incubate tubes at least 5 minutes (maximum is 45 minutes) at room temperature.
4)
Transfer the sample to a cuvette and determine the sample concetration - measure the
absorbance at 595 nm on spectrophotometer (Nano Photometer IMPLEN) and note
down the concentration of the sample in ug/ul. Store this value for next week of
practice.
Products formed in colorimetric reaction absorb light at this wave length. The values of
absorbance correspond to the amount of proteins present in the sample.
Spectrophotometer software calculates sample concentration from sample absorbance
using the calibration equation that was determined previously using several samples
containing BSA in known concentration and stored in the device for later use. For
calculation of undiluted tissue sample concentration the value displayed by the
spectrophotometer has to be multiplied by 10 because of dilution of the sample that was
done to fit the calibration curve range.
Cell and Molecular Biology
Summer semester practice, part I
SOLUTIONS USED
Lysis buffer:
10 mM
100 mM
1 mM
0.5 %
Tris-HCl, pH 8
NaCl
EDTA
SDS (sodium dodecylsulphate)
Store at room temperature.
PBS (phosphate buffered saline):
137 mM
2.7 mM
8.0 mM
1.5 mM
NaCl
KCl
Na2HPO4
K2HPO4
Adjust pH to 7.4, store at 4°C.
Mounting medium with DAPI:
2.44 g
6 ml
6 ml
12 ml
5 g
mowiol
glycerine
distilled water
250 mM Tris-HCl
DAPI (4',6-diamidino-2-phenylindole)
Adjust pH to 8.7, store at 4°C.
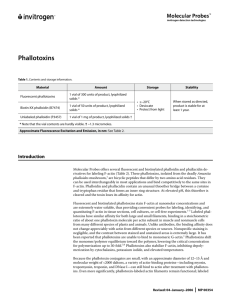
Phalloidin-TRITC and DNase I-Alexa Fluor staining solution:
0.2 g/ml Phalloidin-TRITC and 5 g/ml DNase I solution in PBS, store at 4°C.
Formaldehyde solution with Triton X-100
3.7 % formaldehyde and 0.1% Triton X-100 in PBS, store at 4°C.
 0
0