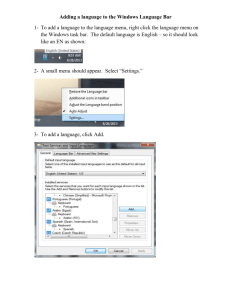
Laboratory 10 (WORD)
advertisement
")
Laboratory 10
Seed Plant Phylogeny (week two)
Compare your trees with the sample trees that your teaching fellow has handed out in lab this week.
Make sure that you understand how the character changes marked on the trees yield the character states
listed in the 6-character data table. Also make sure you understand how the tree length (total number
of character-state changes) was computed. Finally, locate the character reversals (from 1 to 0) and
parallelisms (two or more identical changes of the same character, either 0 to 1 or 1 to 0) on the three
trees.
IV. Building the Full Tree via Computer and Manipulating It to Test Ideas.
Consider the following: the trees you have just finished looking at are for eight groups of organisms
based on six characters. The most recent analysis of the seed plants, done by Kevin Nixon and
coworkers (Nixon et al., 1994) included 103 characters for 14 living groups and 14 extinct groups - a
data table of 103 x 28 cells! No one in her right mind would attempt to infer a phylogeny (build a tree)
from the huge pile of data in this data set by hand. What we do instead is leave it to the computers to
do the job. You still must study the plants and collect the character states, build the data table, and tell
the computer how to do the analysis - but most of the time is in finding the shortest tree, and this
process the computer can do.
In this exercise we will use Nixon’s data set, though only subset of the characters and only for the
living seed plants, to infer phylogenies (that is build trees that hypothesize evolutionary history) using
computer programs. Though there is an array of available programs, most do basically the same thing they build networks based on similarities, then root the trees using the outgroup criterion (that is, a
character shared by the outgroup and some of the members of the study group is primitive, see. p. 8).
For this exercise, we will use two different programs, because they each have different capacities: the
programs are ---- PAUP (stands for Phylogenetic Analysis Using Parsimony) - to find the tree with the fewest
character-state changes (most parsimonious tree).
-- MacClade - to 1) show the tree, 2) learn about the character distributions on the shortest tree, and 3)
consider alternative trees that are longer but assume possibly more appealing evolutionary histories.
A. Finding the Shortest Tree using PAUP
For this part of the exercise, you need to get used to the MacIntosh computers in the computer lab in
the cellar of Marsh Life Science Building. All the files you need should be in your folder - but make
sure you have them, they are three A copy of PAUP 3.1.1
A copy of MacClade version 3.01
A copy of the seed plant data file, “seedplant.dat”
Okay, let’s begin.
1. Open the file called “seedplant.dat.” You may get a message that says the file is locked and
asking you if you want to open it anyway. Answer yes. Since it is a PAUP file, the computer
will open PAUP, then open your data file. Now, choose Save as... from the File pull-down
menu and follow the hints to give the file a name, which makes your own copy of the data file.
Give it any name you like, but we’ll call this file “yourname.dat” from now on. Save your
file on the desktop.
(If you are not used to MacIntoshes, the way to do this is to 1) move the arrow to the word
“File” in the white ribbon at the top of the screen, 2)move the arrow down to the word “Save
as...”, and 3) let go. The computer will guide you from here on.)
2. Now look at the structure of the file. The design of this file is a consequence of the
programmer’s approach to inferring phylogenies using a computer and the particular
programming language he chose. Let’s take a tour of the file.
a. At the top of the file is the file-type label, “#NEXUS”. The third line, “BEGIN
DATA;” is a signal to the computer that the data are about to be fed to it. Then,
under “DIMENSIONS”, the file describes the number of groups we are going to
provide data for (NTAX=11), then the number of characters provided for each of
these groups (NCHAR=103).
b. Then, in the fifth line, comes some language defining characters in the data table.
These are useful to understand.
FORMAT MISSING=? --- If you look at the MATRIX (data table) below, you will
see a number of question marks. These may mean one of three things:
i. truly missing data (for instance if no one has ever studied that part of the
plant)
ii. data missing because a structure has not been invented by a group (for
instance fruit structure for a pine tree makes no sense because pines don’t
have fruits.)
iii. in the case of our data (from Nixon et al., 1994) a character that
- varies within a group
- is confusing as to its homology (for instance it may be unclear exactly
what a leaf is in a group)
GAP=. doesn’t apply to this data set.
SYMBOLS= “0,1,2,3” defines the numbers to be used to represent character states. In this larger data
set some characters have more than two character states. For instance, consider character 27, vein
orders. This character represents the number of times that veins branch, and the number varies. Nixon
et al. chose to divide the character into three character states - not branched, branched once, and
branched twice or more. In the matrix these character states are represented by 0,1, and 2.
The vein-order number character also introduces the idea of defining primitive versus derived character
states. This particular character is considered as “ordered”, that is unbranched veins are primitive,
singly branched veins are derived from unbranched veins, and the more complex branching(branched
twice or more) is in turn derived from the singly branched veins.
Sometimes, scholars do not want to define character states as primitive or derived, in which case the
character is labeled “unordered”.
c. Next comes the MATRIX command, which tells the computer that the data are
next in the file. It ignores the numbers in brackets, i.e. [10
20...], which are
there to help you tell which character number you are looking at in the matrix below.
The matrix itself includes a number or a question mark for each character for each of
the eleven living groups (evolutionary lineages) we are asking the program to build a
tree for.
d. Down at the bottom of the file is a block of commands under “BEGIN
ASSUMPTIONS”. These tell the computer about our particular choices for
analyzing these data.
i. TYPESET * mixed distinguishes between the unordered and ordered
characters in the data set. These are Nixon et al.’s opinions about which is
which, from the original article.
ii. EXSET * exclude is the list of characters we are excluding in this
exercise. These are removed so that just enough characters (32) are
included to give us the same answer as all 103 characters, and it will make it
simpler for you to deal with the trees when you make them.
The final characters are ------- {13 17 20 22 33-35 38-40 47 57-61 64 65 70 71 73 74
83-92} --- Here are the characters and their character states:
CHAR.
13.Vessels
17.Lignin Subunits
20.Resins
22.Leaf Base
33.Stomates
34.Astrosclereids
in
Leaf
35.Strobili
38.Microsporophylls
39.Microsporophylls
40. Microsporangia
per unit
47.Leptomate
Aperture
57.Microgametophyte
58.Pollen Tube
59.Ramiform Pollen
Tubes
60.Stalk Cell
61.Sperm
64.Woody Cones
65.Compound Cone
Units
70.Ovules
71.Micropyle
73.Ovule Growth
74.Outer Seed
Envelope
83.Megaspore Tetrad
84. Megaspore Wall
85.Megagametophyte
86.Megagametophyte
87.Archegonia
88.Egg
89.Early Embryogeny
90.Embryo Maturity
91.Embryo Feeder
92.Seed Germination
KIND
additive
additive
nonadditive
STATE 0
absent
vanillan
absent
simple
haplocheilic
STATE 2
absent
STATE 1
present
syringal groups
present
sheathing
some or all
syndetocheilic
present
unisexual
bisexual
functionally
unisexual
spiral
free
many
whorled/opposite
basally fused
1-4
absent
present
more than
fournucleate
suspended
absent
4-nucleate
present
flagellate
absent
many
absent
nonflagellate
present
few
orthotropous
normal
pachychalazal
absent
anatropous
tubular
endochalazal
present
tetrahedral
thick
monosporic
alveolar
present
cellular
free-nuclear
postshed
absent
cryptocotylar
linear
thin/absent
tetrasporic
nonalveolar
absent
free-nuclear
cellular
preshed
present
phanerocotylar
3-nucleate
penetrating
present
isobilateral
iii. ANCSTATES allzero = 0:ALL. This is the way in which we actually
root the tree, in this case by defining all the zero character state in the data
table as most primitive. Nixon et al. decided on which character states were
most primitive by the outgroup-comparison method we discussed last week,
and coded the character states so that 0 is most primitive for each one..
3. It’s time to run the program. Choose the File pulldown menu from the white bar at the top
of the screen and select Execute yourname.dat to run PAUP, the program that will build
your phylogenetic tree for you, using your file.
a. PAUP will first read the data for our 11 groups using the 32 selected characters.
PAUP reports on its work. You should see the following messages:
Processing of file “yourfile.dat nexus” begins...
Data matrix has 11 taxa, 103 characters
Valid character-state symbols: 0123
Missing data identified by ‘?’
Gaps identified by ‘-’, treated as “missing”
Processing of “yourfile.dat nexus” completed.
PAUP counts all the characters in the data file to come up with 103, but will only use
32, because of the EXSET * exclude command we included. Taxa are our groups.
b. Next we need to ask PAUP to search for the shortest tree. From the Search pull-down
menu choose Heuristic. This sort of search for trees is the fastest, but it may not find the
shortest tree of all - we choose this kind of search to save you time.
The next menu that appears offers you the options that go with the heuristic search. Simply
tap return, since we‘ll use all the default options. PAUP then does the search, and you get two
reports, one about the details of the parameters for the search and a second that reports on the
results of the search, with the command close in a button.
Notice that the program tried 416 rearrangements and found one shortest tree that is 37 steps
long.
Now go ahead and click on the close button.
c Now choose the Trees menu and select Show Trees...and tap return to view a primitive
version of the shortest tree in the PAUP windowframe now. But to really play with this tree,
the other program, MacClade, is better. So save this tree - choose Trees again and select Save
trees to file... at the very bottom of the pull-down menu The name of your PAUP tree file
will be yourfile.dat.trees unless you change it.
Now quit PAUP by choosing the File menu and select the Quit option at the very bottom.
B. Now work with the MacClade program to understand the structure of your phylogeny (hypothesis of
evolutionary history) for the living seed plants. Find the MacClade program and click on it twice to
start it running.
1. Choose the File menu and select Open File... Click on the Desktop button. Now choose the
data file you named, for example “Yourfile.dat” (not the trees file). MacClade and PAUP
recognize each other’s files, so the file will open right up. This time, the data are really easy
to see. The 11 groups are named along the left side in the first column, and the character
states for each character are listed in the column under each character number. Scan across
the right to view all 103 characters, but remember - we are only using the 32 above.
2. To see how MacClade excludes characters, choose the Display menu and select Character
Status. This command reveals a table of characters with information about each. You can
quickly see which characters are included and excluded. Close this window when you’re
done.
3. Now it’s time to open the tree file using MacClade. Once again choose the Display menu,
but this time select Go To Tree Window. You will get a message that says that there are no
tree files stored for this data file - choose the button that says “open tree file”.
A menu appears that allows you to choose your tree file, for instance “yourfile.dat.trees”.
Click on the file and then tap the button to choose it (You can also click twice on the file
name).
A new menu appears with a list of files, including 1.PAUP.1 - this is the tree you created
using PAUP. Select it and tap return. The tree itself will finally appear. Now you can get
down to playing with the tree.
4. Having gotten your tree ready to manipulate, there are three basic things to do.
First, trace a single character’s history and show the outcome in the character states typical of
each group, and
Second, customize the tree to show the character changes,
Third, change the tree around to understand the effects of choosing a different history for the
evolutionary groups.
But first, a simple lesson. Choose the Tools menu. Choose the symbol that looks like this:
This tool allows you to rotate the two branches at a node - try it out. Move the cursor on to
the tree image and click on a line bearing two branches. You will see the branches rotate.
One thing critical to figure out is that either way the tree is telling you the same thing - that the
two branches represent the two evolutionary descendants of one common ancestor.
a.Tracing the History of Single Characters
For this section, here are two characters from the data table for reference.
Character
13.Vessels
20.Resins
0
absent
absent
1
present
present
i. Choose Trace Character from the Trace menu. The changes in
character 13 (vessels) are shown in color, with yellow primitive and blue
derived. (You may want to change the shape of the tree to see it more
easily, now that the character changes are not being displayed.)
ii. Now choose Data Boxes from the Display menu. This box shows the
actual data for each of our eleven living seed plant groups going across the
screen. Scroll up the data in the box until character 13 sits just on top of the
tree - and you should see a combination of the data for character 13 and the
inferred evolutionary history of the character states.
What you see is that Ephedra, Gnetum, Welwitschia, and the angiosperms
all share a character state (vessels present) whereas the rest of the groups
share a character state with our outgroup, the cycads (vessels absent).
Vessels absent is thus the primitive character state (indicated by yellow),
and vessels present is derived (indicated by blue). You can see from the
coloring of the tree that the common ancestor of these four groups is
supposed to have invented vessels (the common ancestor is the line ancestral
to all; it shows the transformation “13:0->1”.)
Understanding this last paragraph is the most important thing you can do in
this lab - it is an illustration of the way in which modern phylogenetic
analysis infers common ancestors based on shared, derived character states.
iii. Look at other characters to see similar histories inferred from character
distributions. Especially look at character 20 -- resins present versus absent
-- since this character helps define a common ancestor for all of the conifers.
b. Displaying Character Changes on the Trees. Now it’s time to use MacClade to
label the tree with the character changes inferred by PAUP.
i. Go to the Trace menu and choose All Changes Options. Here you will
be presented with a box that customizes the tree. Choose...
---”almost all possible changes”
---choose “Trace All” down at the bottom right. (This can also be done
from the Trace menu by selecting Trace All Changes.)
ii. Go to the Display menu and choose Trace Labeling.
---Click on the symbol labeled “label by characters changing”
---Check the box labeled “Show states changing”.
iii. Now, looking at the tree, you will see that the character changes are
impossible to read. Go back to the Tree Size and Shape under the Display
menu and choose four times taller than wide. Also for tree shape choose the
square corner option. This should open up the tree so you can see all the
characters changing.
Notice that most of the character changes make sense. You can see the
character number followed by the change, for instance” 91:0->1”. In some
cases, what you see is the following: :71:0/1->1”. These are cases in which
the simplest solution does not include a decision between two character
states for a particular place in the tree (both options yield the same tree
length). 71:0/1->1 translated means : “For character 71 at this internode, it is
just as parsimonious for the ancestor to have character 0 as character 1, take
your choice.” You can prove this yourself by counting up the number of
character-state changes under each assumption. This is best don eby
sketching the tree twice, once for parallel
iv. Look at the two characters, 13 and 20. You should see that MacClade
shows them transforming from primitive to derived at the expected places.
Vessels originate (13:0->1) in the common ancestor of Ephedra, Gnetum,
Welwitschia, and the angiosperms. Resins are invented (20:0->1) in by the
common ancestor of the conifers.
v. If you can, you may choose to print this tree as it now stands. Printing is
done from the File menu, by selecting Print. You will have to choose
among several options on the Print menu.
c. Manipulating the Tree to Test Alternative Hypotheses.
i. Remember the woody cone character? Here it is again:
Character
64.Woody Cones
0
absent
1
present
Some specialists have suggested that the podocarps and yews, which look
like conifers but don’t have cones, have lost their cones (a reversal of
character 64) instead of never having invented them. Use the Trace
Character command to show the character-state distribution and inferred
history of this trait (choose character 64 from the box at the bottom right).
How many more changes would be required in the tree for the common
ancestor of yews, podocarps and conifers to have invented woody cones?
(You must figure this out by looking at the character state changes on the
current tree and figuring out how many more changes are needed with this
new constraint.)
ii. Some specialists have felt that the angiosperms were most closely related
to the genus Gnetum among seed plants. How does the length of the tree
change if you move the angiosperms into a position where they and Gnetum
arise from the same common ancestor?
To answer this question - Choose the arrow tool from the Tool menu if you don’t have the arrow.
- Move the arrow to the line leading to Gnetum.
- Click and hold down while you move the arrow to the line leading to the
angiosperms.
- Let go.
MacClade will rebuild the tree with Gnetum and the angiosperms as
descendants of a single common ancestor (as sister groups) and report the
total length of the tree down at the bottom right in the box.
Adjust the tree size and shape to 4 times normal size and 1 times as high as
wide. Move the cursor to show just the following three groups angiosperms, Gnetum, and Welwitschia. Count the number of parallelisms
and reversals on the modified tree (with Gnetum and the angiosperms as
sister groups) and on the original tree (with Gnetum and Welwitschia as
sister groups). These counts will demonstrate to you how trees get longer
and shorter.
Look at the tree length reported in the PAUP 1 Box: it has gone from 37 to
43 steps.
There is an additional problem with the hypothesis that Gnetum is the sister
group of the angiosperms. Can you figure out what it is?
V. Considering a Molecular Data Set (Albert et al., 1994) for the Same Taxa (Not
Written 1999)
RELEVANT LITERATURE
Albert, V.A., A. Backlund, K. Bremer, M.W. Chase, J.R. Manhart, B.D. Mishler, and K.C. Nixon.
1994. Functional constraints and rbcL evidence for land plant phylogeny. Annals Missouri
Botanical Garden 81:534-567. [shows trees based on integrated molecular and morphological
data for both all and only living taxa]
Doyle, J.A. and M. J. Donoghue. 1986. Seed plant phylogeny and the origin of the angiosperms:
an experimental cladistic approach. Botanical Review 52(4):321-431.
Doyle, J.A., M.J. Donoghue, and E.A. Zimmer. 1994. Integration of morphologiccal and
ribosomal DNA data on the origin of angiosperms. Annals Missouri Botanical Garden 81:
419-450.
Nixon, K.C., W.L. Crepet, D. Stevenson, and E.M. Friis. 1994. A reevaluation of seed plant
phylogeny. Annals Missouri Botanical Garden 81:484-533. [source of the data for this lab]