Splitting the dataset into two parts
advertisement

Data Mining for Prediction of Genes Associated with a given Biological Function
William Perrizo
ABSTRACT:
Comprehensive Genome Databases provide several catalogues of information on some genomes
such as yeast (Saccaromyces cerevesiae). We show the potential for using gene annotation data, which
includes phenotype, localization, protein class, complexes, enzyme catalogues, pathway information,
and protein-protein interaction, in predicting the functional class of yeast genes. We predict a rank
ordered list of genes for each functional class from available information and determine the relevance of
different input data through systematic weighting using a genetic algorithm. The classification task has
many unusual aspects, such as multi-valued attributes, classification into overlapping classes, a
hierarchical structure of attributes, many unknown values, and interactions between genes. We use a
vertical data representation called the P-tree representation, that treats each domain value as an attribute.
The weight optimization uses ROC evaluation in the fitness measure to handle the sparseness of the
data. We include the number of interactions as a prediction criterion, based on the observation that
important genes with many interactions are more likely to have some function. The result of the weight
optimization confirms this observation.
Keywords: data mining, prediction, classification, function, similarity, genetic algorithms, P-tree
1. INTRODUCTION
During the last three decades, the emergence of molecular and computational biology has shifted
classical genetics research to the study at the molecular level of the genome structure of different
organisms. As a result, the rapid development of more accurate and powerful tools for this purpose has
produced overwhelming volumes of genomic data that is being accumulated in more than 300 biological
databases. Gene sequencing projects on different organisms [1] have helped to identify tens of thousands
of potential new genes, yet their biological function often remains unclear. Different approaches have
been proposed for large-scale gene function analysis. Traditionally functions are inferred through
sequence similarity algorithms [2] such as BLAST or PSI-BLAST [3]. Similarity searches have some
shortcomings. The function of genes that are identified as similar may be unknown, or differences in the
sequence may be so significant as to make conclusions unclear. For this reason some researchers use
sequence as input to classification algorithms to predict function [4]. Another common approach to
function prediction uses two steps. Genes are first clustered based on similarity in expression data, and
then clusters are used to infer function from genes in the same cluster with known function [5].
Alternatively function has been directly predicted from gene expression data using classification
techniques such as Support Vector Machines [6].
We show the potential for the use of gene annotation data, which includes phenotype,
localization, protein class, complexes, enzyme catalogues, pathway information, and protein-protein
interactions, in predicting the functional class of yeast genes. Phenotype data has been used to construct
individual rules that can predict function for certain genes based on the C4.5 decision tree algorithm [7].
The gene annotation data we used was extracted from the Munich Information Center for Protein
Sequences (MIPS) database [8] and then processed and formatted in a way that fits our purpose. MIPS
has a genomic repository of data on Saccharomyces cerevesiae (yeast). Functional classes from the
MIPS database include among many others, Metabolism, Protein Synthesis and Transcription. Each of
these is in turn is divided into classes and then divided again into subclasses to yield a hierarchy of up to
five levels. Each gene may have more than one function associated with it. In addition MIPS has
catalogues with other information such as phenotype, localization, protein class, protein complexes,
enzyme catalogue and data on protein-protein interactions. In this paper, we predict function for genes at
the highest level in the functional hierarchy. Despite the fact that yeast is one of the most thoroughly
studied organisms, the function of 30 – 40 % of its ORFs remain currently unknown. For about 30% of
the ORFs no information whatsoever is available, and for the remaining ones unknown attributes are
very common. This lack of information creates interesting challenges that cannot be addressed with
standard data mining techniques. We have developed novel tools with the hope of helping biologists by
providing experimental direction concerning the function of genes with unknown functions.
2. EXPERIMENTAL METHOD
Gene annotations are untypical data mining attributes in many ways. Each of the MIPS
catalogues has a hierarchical structure. Each attribute, including the class label, gene function, is
furthermore multi-valued. We therefore have to consider each domain value as a separate binary
attribute rather than assigning labels to protein classes, phenotypes, etc. For the class label this means
that we must classify into overlapping classes, which is also referred to as multi-label classification,
rather than multi-class classification in which the class label is disjoint. To this end we represent each
domain value of each property as a binary attribute that is either one (gene has the property) or zero
(gene doesn't have property). This representation has some similarity to bit-vector representations in
Market Basket Research, in which the items in a shopping cart are represented as 1-values in the bitvector of all items in a store. The classification problem is correspondingly broken up into a separate
binary classification problem for each value in the function domain. The resulting classification problem
has more than one thousand binary attributes, each of which is very sparse. Two attribute values should
be considered related if both are 1, i.e., both genes have a particular property. Not much can be
concluded if two attribute values are 0, i.e., both genes do not have a particular property.
Classification is furthermore influenced by the hierarchical nature of the attribute domains. Many
models exist for the integration of hierarchical data into data mining tasks, such as text classification,
mining of association rules, and interactive information retrieval, among others[19],[20],[21],[22].
Recent work [17] introduces similarity measurements that can exploit a hierarchical domain, but focuses
on the case where matching attributes are confined to be at the leaf level of the hierarchy. The data set
we consider in this paper has poly-hierarchical attributes, where attributes must be matched at multiple
levels. Hierarchical information is represented using a separate set of attribute bit columns for each level
where each binary position indicates the presence or the absence of the corresponding category.
Evidence for the use of multiple binary similarity metrics is available in the literature and usage is based
on the computability and the requirement of the application [25]. In this work we use a similarity metric
identified in the literature as “Russel-Rao” and the definition is given bellow[25].
Given 2 binary vectors Zi and Zj with N dimensions (categorical values)
N 1
similarity ( Zi, Zj )
Z
k 0
i ,k
Z j ,k
N
The above similarity measure will count the number of matching bits in the two binary vectors and
divide by the total number of bits. A similarity metric is defined as a function that assigns a value to the
2
degree of similarity between object i and j. For each similarity measure the corresponding dissimilarity
is defined as the distance between two objects and should be non-negative, commutative, adhere to
triangle inequality, and reflexive to be categorized as a metric. Dissimilarity functions that show partial
conformity to the above properties are considered as pseudo metrics. The corresponding dissimilarity
measure only shows the triangular inequality when M<N/2 where N is the number of dimensions and M
is the max(X.Y). For this application we find the use of the above as appropriate. It is also important to
note that in this application the existence of a categorical attribute for a given object is more valuable
than the nonexistence of a certain categorical attribute. In other words “1” is more valuable than a “0” in
the data. So the count of matching “1” is more important for the task than the count of matching “0”.
The ptree data structure we use also allows us to easily count the number of matching “1”s with the use
of a root count operation.
Similarity is calculated considering the matching similarity at each individual level. The total
similarity is the weighted sum of the individual similarities at each level on the hierarchy. The total
weight for attributes that match at multiple levels is thereby higher indicating a closer match. Counting
matching values corresponds to a simple additive similarity model. Additive models are often preferred
for problems with many attributes because they can better handle the low density in attribute space, also
referred to as "curse of dimensionality" [9].
2.1 Similarity & Weight Optimization
Similarity models that consider all attributes as equal, such as K-Nearest-Neighbor classification
(KNN) work well when all attributes are similar in their relevance to the classification task. This is,
however, often not the case. The problem is particularly pronounced for categorical attributes that can
only have two distances, namely distance 0 if attributes are equal and 1 or some other fixed distance if
attributes are different. Many solutions have been proposed, that weight dimensions according to their
relevance to the classification problem. The weighting can be derived as part of the algorithm [10]. In
an alternative strategy the attribute dimensions are scaled, using, e.g., a genetic algorithm, to optimize
the classification accuracy of a separate algorithm, such as KNN [11]. Our algorithm is similar to the
second approach, which is slower but more accurate. Modifications were necessary due to the nature of
the data. Because class label values had a relatively low probability of being 1 we chose to use AROC
values instead of accuracy as criterion for the optimization [12]. Nearest neighbor evaluation was
replaced by the counting of matches as described above. We furthermore included importance measures
into the classification that are entirely independent of the neighbor concept. We evaluate the importance
of a gene based on the number of possible genetic and physical interactions its protein has with the
proteins of other genes. Interactions with lethal genes, i.e., genes that cannot be removed in gene
deletion experiments because the organism cannot survive without them, were considered separately.
The number of items of known information, such as localization and protein class, was also considered
as importance criterion.
2.2 ROC Evaluation
Many measures of prediction quality exist, with the best-known one being prediction accuracy.
There are several reasons why accuracy is not a suitable tool for our purposes. One main problem
derives from the fact that commonly only few genes are involved in (positive) for a given function. This
leads to large fluctuations in the number of correctly predicted participant genes (true positives).
Secondly we would like to get a ranking of genes rather than a strict separation into participant and nonparticipant since our results may have to be combined with independently derived experimental
probability levels. Furthermore we have to account for the fact that not all functions of all genes have
3
been determined yet. Similarly there may be genes that are similar to ones that are involved in the
function, but are not experimentally seen as such due to masking. Therefore it may be more important
and feasible to recognize a potential candidate than to exclude an unlikely one. This corresponds to the
situation faced in hypothesis testing: A false negative, i.e., a gene that is not recognized as a candidate,
is considered more important than a false positive, i.e., a gene that is considered a candidate although it
isn't involved in the function.
The method of choice for this type of situation is ROC (Receiver Operating Characteristic)
analysis [12]. ROC analysis is designed to determine the quality of prediction of a given property, such
as a gene being involved in a phenotype. Samples that are predicted as positive and indeed have that
property are referred to as true positive samples, samples that are negative, but are incorrectly classified
as positive, are false positive. The ROC curve depicts the rate of true positives as a function of the false
positive rate for all possible probability thresholds. A measure of quality of prediction is the area under
the ROC curve. Our prediction results are all given as values for the area under the ROC curve
(AROC). To construct a ROC curve samples are ordered in decreasing likelihood of being positive.
The threshold that delimits prediction as positive is then continuously varied. If all true positive samples
are listed first the ROC curve will start out by following the y-axis until all positive samples have been
plotted and then continue as horizontal for the negative samples. With appropriate normalization the
area under this curve is 1. If samples are listed in random order the rate of samples that are true positive
and ones that are false positive will be equal and the ROC curve will be a diagonal with area 0.5. The
following table gives some examples of ordered samples and the respective AROC value.
Example with Area under the
ROC curve = 1
TRUE
TRUE
FALSE
FALSE
FALSE
FALSE
Example with Area under the
ROC curve = 0.625
FALSE
TRUE
FALSE
TRUE
FALSE
FALSE
Example with Area under the
ROC curve = 0.5
FALSE
TRUE
FALSE
FALSE
TRUE
FALSE
Table 1 Example ROC curve values.
2.3 Data Representation
2.3.1 P-TREE1 VERTICAL DATA REPRESENTATION
The input data was converted to P-trees [13], [14], [15], [16]. P-trees are a lossless, compressed,
and data-mining-ready data structure. This data structure has been successfully applied in data mining
applications ranging from Classification and Clustering with K-Nearest-Neighbor, to Classification with
Decision Tree Induction, to Association Rule Mining for real world data [13], [14], [15]. A basic P-tree
represents one attribute bit that is reorganized into a tree structure by recursively sub-dividing, while
recording the predicate truth value regarding purity for each division. Each level of the tree contains
truth-bits that represent pure sub-trees and can then be used for fast computation of counts. This
construction is continued recursively down each tree path until a pure sub-division is reached that is
entirely pure. The basic and complement P-trees are combined using Boolean algebra operations to
1
P-tree technology is patent pending, This work is partially supported by GSA Grant ACT#: K96130308.
4
produce P-trees for values, entire tuples, value intervals, or any other attribute pattern. The root count of
any pattern tree will indicate the occurrence count of that pattern. The P-tree data structure provides the
perfect structure for counting patterns in an efficient manner. The data representation can be
conceptualized as a flat table in which each row is a bit vector containing a bit for each attribute or part
of attribute for each gene. Representing each attribute bit as a basic P-tree generates a compressed form
of this flat table.
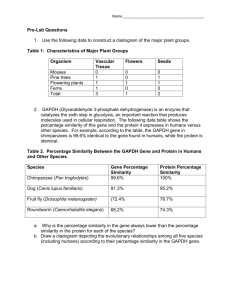
The following figure shows how an attribute from a poly-hierarchical domain is encoded into the
P-tree data structure (bit vector form). A poly-hierarchical attribute groups a set of attributes in a
recursive sub-hierarchy where each node represents an individual attribute from the attribute domain and
no restrictions are imposed on the number of nodes or number of levels. In our representation the
hierarchy is encoded in a depth first manner. The level boundaries with respect to the P-tree index are
preserved as meta information for the attributes. This information is used in computing the weighted
sum of the similarities between objects.
A0
O(A0, A2, A5, A9)
(b)
A2
A1
A3
A0
A4
A2
A6
A5
O(1, 0, 1, 0, 0, 1, 0, 0, 0, 1)
A5
(c)
A7
A8
A9
A9
(a)
(d)
Figure 1: (a) An example poly-hierarchical attribute domain (b) An example data attribute object drawn
from this domain (c) Bit vector representation of the object. (d) A link that represents the data attribute
object in the hierarchy.
Experimental class labels and the other categorical attributes were each encoded in single bit
columns. Protein-interaction was encoded using a bit column for each possible gene in the data set,
where the existence of an interaction with that particular gene was indicated with a truth bit.
2.4 Implementation
The work presented in this paper required data extraction from the MIPS database, data cleaning,
developing a similarity metric and optimizing the similarity metric. Following figure shows an outline
of the approach.
5
Standard GA
Wi
MIPS
Data
Domain
Knowledge
Train
List
Data Cleaning,
Derived
attributes
Gene
Data
HTML data
extractors
Test
List
ROC
Nearest Neighbor
Predictor
Similarity based on
weighted sum of
matching attributes
and importance
Predictor
with optimized
weights
Final
Prediction
Figure 2 Outline of approach.
Gene data from the MIPS database was retrieved using HTML data extractors. With assistance
from domain knowledge a few tools were developed to clean the data and also extract some derived
attributes as training data. One of the steps we took in data cleaning consisted in extracting the Function
sub-hierarchy “Sub-cellular localization” from the MIPS Function hierarchy and treating it as a property
in its on right. The basis for this step was the recognition that Sub-cellular Localization represents
localization information rather than function information. The information in Sub-cellular Localization
can be seen as somewhat redundant to the separate localization hierarchy. We didn’t explicitly decide in
favor of either but rather left that decision to the weight optimization algorithm. Sub-cellular
Localization could be seen to perform somewhat better than the original localization information,
suggesting that the data listed under Sub-cellular Localization may be cleaner than localization data.
The following equations summarize the computation using P-trees, where Rc: root-count, W: weight,
Px:P-tree for attribute x (Pg is the interaction P-tree for gene g), At: attribute count, ptn: class-partition,
Im: gene-importance, Lth: lethal gene (cannot be removed in gene deletion experiments), g: test gene to
be classified, f: feature, Ip: Interaction property, gn: genetic interaction (derived in vivo), ph:physical
interaction (derived in vitro), lt: lethal, ClassEvl: evaluated value for classification, : P-tree AND
operator.
ClassEvl ptn
f g
(g)=
Wf ×Rc(Pptn Pf
f
{gn,ph,lt}
) +
WIp ×Rc(Pg PIp ) WAt × At (g)
Ip
Equation 1 Prediction function for gene (g)
For each feature attribute (f) of each test gene, g, the count of matching features for the required
partition was obtained from the root-count by ANDing the respective P-trees. We can obtain the number
of lethal genes interacting with a particular gene, g, with one P-tree AND operation. It is possible to
retrieve the required counts without a database scan.
6
Due to the diversity of the function class and the nature of the attribute data, we need a classifier
that does not require a fixed importance measure on its features, i.e., we need an adaptable mechanism
to arrive at the best possible classifier. In our approach we optimize the weight space, W, with a
standard Genetic Algorithm (GA) [23]. The set of weights on the features represented the solution space
that was encoded for the GA. The AROC value of the classified list was used as the GA fitness evaluator
in the search for an optimal solution. AROC evaluation provides a smoother and accurate evaluation of
the success of the classifier than standard accuracy which is vital ingredient for a successful hill climb.
3. RESULTS
We demonstrate the prediction capability of the proposed algorithm, through simulations that
predict different function classes at the highest level in the hierarchy. Class sizes range from small
classes with only very few genes, e.g., Protein activity regulation with 6 genes to large classes such as
Transcription with 769 genes, or more than 10% of the genome. We compare our results with [7] since
this is the most closely related work we have found that uses gene annotation data (phenotype).
Differences with respect to our approach are however such that comparison is difficult. In [7] a set of
rules for function classification is produced and mean accuracies and the biological relevance of some of
the significant rules are reported. The paper does not have the goal of producing an overall high
prediction accuracy, and does therefore not evaluate such and accuracy. Since we build a single
classifier for each function class, we must compare our results with the best rule for each function class.
We were able to download the related rule output files referred in [7] and pick the best accuracy values
recorded for each type of classification. These values were used for the comparison of our classifier. As
discussed previously we strongly discourage the use of standard accuracy calculation for the evaluation
of classification techniques, but use it here for comparison purposes. [7] does not report rules for all the
functional classes we have simulated. ‘ROC’ and ‘Acc’ indicate the accuracy indicators for our
approach for the respective function classes. Accuracy values for the comparative study [7] on
functional classification are given as ‘CK_Acc’. As it can be seen on the following figure for the classes
where the accuracy values are available, our approach performs better in all cases except with
Metabolism.
0.8
ROC
Acc
CK_Acc
Function Class
7
METABOLISM
CONTROL OF CELLULAR
ORGANIZATION
PROTEIN ACTIVITY
REGULATION
CELL FATE
REGULATION OF /
INTERACTION WITH
CELLULAR ENVIRONMENT
CELL CYCLE AND DNA
PROCESSING
CELLULAR
COMMUNICATION/SIGNAL
TRANSDUCTION
MECHANISM
ENERGY
CELLULAR TRANSPORT
AND TRANSPORT
MECHANISMS
TRANSCRIPTION
0
TRANSPORT
FACILITATION
0.4
PROTEIN SYNTHESIS
Accuracy
Classification Accuracy
Figure 3 Shows the Classification accuracy.
As observed in previous work [7], [24], different functional classes are predicted better by different
attributes. This is evident in the final optimized weights given in appendix table A1 for the classifier.
The weights do not show a strong pattern indicating a global set of weights that could work for all the
classifications. Overall the protein-class feature had higher weights in predicting the functions for most
of the functional categories followed by complexes. In protein synthesis functional category the
combined similarity in complexes (weight = 1.0), protein class (weight = 1.033), and Phenotype
(weight=1.2) lead to significant prediction accuracy with AROC = 0.90. Transport facilitation had a high
AROC value of 0.87 from the combined similarity in protein class (weight=1.867), localization
(weight=1.46), and complexes (weight=1.1). This observation highlights the importance of using protein
class and complexes as possible experimental features for biologists in the identification of gene
functions.
We include the number of interactions in such a way that many interactions directly favor
classification into any of the function classes. This contribution to the classification is not directly based
on similarity with training samples, and is thereby fundamentally different from the similarity-based
contributions. This is an unusual procedure that is justified by the observation that it improves
classification accuracy, which can be seen from the non-zero weights for interaction numbers. From a
biological perspective the number of interactions is often associated with importance of a gene. An
important gene is more likely to be involved in many functions than and unimportant one. The weights
in table A1 show that for all function classes interactions benefited classification.
It is worth analyzing if the effect is a simple result of the number of experiments done on a gene.
If many experiments have been done many properties may be known and consequently the probability
of knowing a particular function may be higher. Such an increased probability of knowing a function
would not help us in predicting unknown functions of genes on which few experiments have been done.
To study this we tried including the total number of known items of information, such as the total
number of known localizations, protein classes, etc. as a component. The weights of this component
were almost all zero as can be seen in table A1. This suggests that the number of interactions is indeed a
predictor of the likelihood of a function rather than of the likelihood of knowing about a function.
Use of a genetic algorithm requires the evaluation of the fitness function on the training data.
This requires a data scan for each evaluation. In this work the reported results were obtained by 100x40
evaluations for the genetic algorithm for each function class prediction. With the Ptree based
implementation each function class prediction takes 0.027 milliseconds on a Intel P4 2.4 GHz machine
with 2Gb of memory. The corresponding time for a horizontal implementation is 0.834 milliseconds. In
the horizontal approach the algorithm needs to go through all the attributes of a gene blindly to compute
the matching attribute count for a given gene. In contrast in the vertical approach we can count the
matching attributes by counting only the Ptrees for those attributes that are present for the given gene to
be predicted. The large difference in compute time clearly indicates the applicability of the vertical
approach for this application.
4. CONCLUSION
We were able to demonstrate the successful classification of gene function from gene annotation
data. The multi-label classification problem was solved by a separate evaluation of rank ordered lists of
genes associated with each function class. A similarity measure was calculated using weighted counts of
8
matching attribute values. The weights were optimized through a Genetic Algorithm. Weight
optimization was shown to be indispensable as different functions were predicted by strongly differing
sets of weights. The AROC value was shown to be a successful fitness evaluator for the GA. Results of
the weight optimization can be directly used in giving biologists an indication to what defines a
particular function.
Furthermore, it was interesting to note that quantitative information, such as the number of
interactions, played a significant role. In summary we found that our systematic weighting approach and
P-tree representation allowed us to evaluate the relevance of a rich variety of attributes.
5. REFERENCES
[1] Beck, S. and Sterk, P. Genome-scale DNA sequencing: where are we? Curr. Opin. Biotechnol.
9,116-120, 1998.
[2] Pellegrini,M., Marcotte,E. M., Thompson,M. J., Eisenberg,D. & Yeates,T. O. Assigning protein
functions by comparative genome analysis: protein phylogenetic profiles. Proc. Natl Acad. Sci.
USA 96, 4285-4288, 1999.
[3] Altschul, S. F., Madden, T. L., Shaffer, A. A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.
J.Gapped BLAST and PSI-BLAST: A new generation of protein database search programs, Nucleic
Acids Res. 25, 3389-3402, 1997.
[4] King, R. D., Karwath, A, Clare, A., and Dehaspe, L. The utility of different representations of
protein sequence for predicting functional class. Bioinformatics 17 (5), 445-454, 2001.
[5] Wu, L., Hughes, T. R., Davierwala, A. P. , Robinson, M. D., Stoughton, R., and Altschuler, S. J.
Large-scale prediction of Sccharomyces cerevisiae gene function using overlapping transcriptional
clusters. Nature 31, 255-265, 2002.
[6] Brown, M., Nobel Grundy, W., Lin, D., Cristianini, N., Sugnet, C., Furey, T. S., Ares, M. Jr., and
Haussler, D. Knowledge-based analysis of microarray gene expression data using support vector
machines. Proceedings of the National Acedemy of Sciences USA, 97(1), 262-267, 1997.
[7] Clare, A. and King, R. D. Machine learning of functional class from phenotype data.
Bioinformatics 18(1), 160-166, 2002.
[8] http://mips.gsf.de/
[9] Hastie, T., Tibshirani, R., and Friedman, J. The elements of statistical learning: data mining,
inference, and prediction, Springer-Verlag, New York, 2001.
[10] Cost, S. and Salzberg, S., A weighted nearest neighbor algorithm for learning with symbolic
features, Machine Learning, 10, 57-78, 1993.
[11] W.F. Punch, E.D. Goodman, M. Pei, L. Chia-Shun, P. Hovland, and R. Enbody, Further research
on feature selection and classification using genetic algorithms, Proc. of the Fifth Int. Conf. on
Genetic Algorithms, pp 557-564, San Mateo, CA, 1993.
[12] Provost, F., Fawcett, T.; Kohavi, R., The Case Against Accuracy Estimation for Comparing
Induction Algorithms, 15th Int. Conf. on Machine Learning, pp 445-453, 1998.
[13] Ding, Q., Ding, Q., Perrizo, W., ARM on RSI Using P-trees, Pacific-Asia KDD Conf., pp. 66-79,
Taipei, May 2002.
[14] Ding, Q., Ding, Q., Perrizo, W., Decision Tree Classification of Spatial Data Streams Using Peano
Count Trees, ACM SAC, pp. 426-431, Madrid, Spain, March 2002.
[15] Khan, M., Ding, Q., Perrizo, W., KNN on Data Stream Using P-trees, Pacific-Asia KDD, pp. 517528, Taipei, May 2002.
[16] Perrizo, W., Peano Count Tree Lab Notes, CSOR-TR-01-1, NDSU, Fargo, ND, 2001.
9
[17] Prasanna Ganesan, Hector Graia-Molina, and Jennifer Widom. Exploiting Hierarchical Domain
Structure to Compute Similarity. ACM Transaction on Information Systems, Vol. 21, No. 1,
January 2003, Pages 64-93.
[18] Qin Ding, Maleq Khan, Amalendu Roy and William Perrizo. The P-tree Algebra. Proceedings of
ACM Symposium on Applied Computing (SAC'02), Madrid, Spain, March 2002, pp. 426-431.
[19] Feldman, R. and Dagan, I. 1995. Knowedge discovery in textual databases. In proceeding of KDD95.
[20] Han and Fu Han, J. and Fu, Y. 1995. Discovering the multi-level association rules from large
databases. In Proceeding of VLDB ’95, 420-431.
[21] Scott, S and Matwin, S. 1998. Text classification using WordNet hypernyms., In Proceeding of the
Use of the WordNet in Natural Language Processing Systems. Association for Computational
Languistics.
[22] Srikant, R and Agarwal, R. 1995. Mining generalized association rules., In Proceedings of VLDB
’95 407-419.
[23] Goldberg, D.E., Genetic Algorithms in Search Optimization, and Machine Learning, Addison
Wesley, 1989.
[24] Pavlidis, P., J. Weston, J. Cai, and W. Grundy (2001). Gene functional classification from
heterogeneous data. In Proceedings of the Fifth International Conference on Computational
Molecular Biology (RECOMB 2001).
[25] (J. D. Tubbs. A note on binary template matching. Pattern Recognition, 22(4):359–365, 1989.)
TRANSCRIPTION
CELLULAR TRANSPORT AND
TRANSPORT MECHANISMS
ENERGY
CELLULAR COMMUNICATION/SIGNAL
TRANSDUCTION MECHANISM
CELL CYCLE AND DNA PROCESSING
REGULATION OF / INTERACTION
WITH CELLULAR ENVIRONMENT
CELL FATE
PROTEIN ACTIVITY REGULATION
CONTROL OF CELLULAR
ORGANIZATION
METABOLISM
Location
Subcelular Location
Protein Class
Protein Complex
Pathway
Enzyme Catalogue
Phenotype
Lethal Interactions
Genetic Interactions
Physical Interactions
TRANSPORT FACILITATION
\ Function
Weight Attributes \
PROTEIN SYNTHESIS
APPENDIX
0.13
0.13
0.93
0.30
1.10
0.60
0.30
0.00
0.10
1.20
0.27
1.47
2.00
0.60
0.70
0.70
1.50
0.00
1.10
1.20
0.13
0.27
1.47
1.40
1.00
0.00
1.50
0.80
1.40
0.60
0.00
0.00
1.20
1.20
1.40
0.60
1.50
0.00
0.00
0.60
0.93
0.40
0.40
1.30
1.00
1.20
1.00
0.00
0.90
1.30
0.00
0.00
2.00
0.40
0.40
0.80
0.80
0.10
0.20
1.40
0.13
0.00
1.20
1.40
0.70
1.20
0.50
0.10
0.80
1.30
0.00
0.00
1.60
1.30
1.40
0.10
0.00
0.00
0.50
0.80
0.00
0.00
1.60
1.30
1.50
0.00
1.30
0.00
1.10
1.20
1.33
1.87
2.00
1.50
0.90
0.10
0.00
1.00
0.40
1.30
0.13
0.00
0.80
0.10
0.30
0.90
0.50
0.00
1.00
1.40
1.60
0.27
0.00
1.40
0.10
1.20
0.70
0.00
1.00
1.20
10
Protein-Protein Interaction 0.10 0.00
Attribute Count 0.00 0.00
Lethal Gene 1.30 0.00
1.20
0.00
1.50
1.20 0.20
0.00 0.00
1.10 0.30
0.40 0.80 1.00 0.10
0.00 0.00 0.00 0.00
0.30 0.50 0.10 0.60
0.00
1.50
0.00
1.20 0.20
0.00 0.00
0.60 0.00
Prediction Accuracy :ROC 0.895 0.873 0.836 0.752 0.679 0.677 0.675 0.623 0.554 0.503 0.4581 0.428
Number of Genes in Class 331 312 769 184
78
20 284 106 158
6
89 750
Table A1: Attribute Based convergent weights from the Genetic Algorithm Optimization Step.
11