polb23577-sup-0001-suppinfo01

Supporting Information for
Crystallization and Orientation of Isotactic
Poly(propylene) in Cylindrical Nanopores
Dariya K. Reid a , Bridget A. Ehlinger a , Lin Shao b , and Jodie L. Lutkenhaus a* a Artie McFerrin Department of Chemical Engineering, Texas A&M University, College
Station, Texas, 77843 b Department of Chemical & Environmental Engineering, Yale University, New Haven,
Connecticut 06511, USA
*Corresponding Author: e-mail jodie.lutkenhaus@che.tamu.edu
Avrami Kinetics
Figure S1. Avrami Schematic
Figure S2. Relative Crystalline Fraction
S2
S2
S3
S4 Table S1. Avrami Parameters
Table S2. Values for 2θ of (hkl) reflections for the α-phase of bulk iPP, iPP-200, iPP-40 and iPP-15
Recrystallization of iPP
Figure S3. DSC of Recrystallized iPP
Experimental
Figure S4. Experimental setup for XRD
Removal of Bulk Surface Layer
Figure S5. DSC of Plasma-Etched iPP in AAO
S9
S9
References S10
S5
S6
S6
S7
S9
S1
Avrami Kinetics
Figure S1 schematically shows the method used to analyze the isothermal crystallization data. The initial point of crystallization, also labeled as τ = 0 or A, is the point when the data initially deviates from the baseline after the equilibrium isothermal temperature has been reached. The end of crystallization or infinite crystallization, B, is some small time after the baseline has been reached. Additionally, time-zero was defined as the point when the isothermal temperature was initially reached during the rapid temperature drop, see Figure 3 of paper.
Figure S1. Schematic representation of cursor placement for relative crystalline fraction integration.
S2
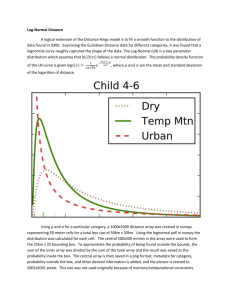
Figure S2.
The evolution of the relative crystalline fraction X(τ) , obtained using equation
1, as a function of τ for (a) bulk-iPP, (b) iPP-200 and (c) iPP-40. Data were taken from
Figure 3 and Eqn 1.
S3
Table S1. Avrami parameters for bulk iPP and iPP infiltrated into AAO templates of varying pore diameter crystallized at varying temperatures ( T c
).
Samples T c
( o C) t
1/2
(min)
Range n
Bulk-iPP 128 1.71 0.031 - 0.407 2.15±0.02
129 2.53 0.028 - 0.404 2.18±0.01 k × 100
(min
-n
)
25.6±0.2
10.93±0.06
R
2
0.99954
0.99960
130 3.69 0.032 - 0.403 2.127±0.006 4.88±0.02 0.99982
131 5.50 0.029 - 0.398 2.190±0.003 1.773±0.006 0.99994
132 8.36 0.029 - 0.402 2.178±0.002 0.697±0.002 0.99997
133 12.49 0.031 - 0.399 2.107±0.002 0.329±0.002 0.99990
134 18.76 0.030 - 0.401 2.021±0.002 0.1789±0.0006 0.99993
135 26.68 0.030 - 0.400 1.953±0.001 0.1117±0.0002 iPP-200 126 4.95 0.029 - 0.401 1.634±0.005 5.63±0.02
127 6.07 0.030 - 0.400 1.645±0.006 4.02±0.02
0.99999
0.99976
0.99964
128 7.74 0.030 - 0.398 1.620±0.005 2.88±0.02
129 8.91 0.030 - 0.400 1.630±0.004 2.20±0.01
0.9996
0.99977
130 11.02 0.029 - 0.399 1.620±0.003 1.585±0.007 0.99982
131 13.43 0.031 - 0.400 1.587±0.002 1.228±0.004 0.99992
132 17.52 0.030 - 0.401 1.571±0.001 0.851±0.002 0.99993
133 21.42 0.030 - 0.400 1.570±0.001 0.613±0.001 0.99995 iPP-40
0.028 - 0.048
1.62±0.01
3.503±0.005
0.99991
132
133
135
7.28
10.88
15.03
0.052 - 0.398
0.102 - 0.401
0.097 - 0.399
1.502±0.006
0.030 - 0.099 1.72±0.01
1.437±0.005
1.376±0.004
3.83±0.03
1.80±0.01
2.47±0.02
0.029 - 0.095 1.536±0.009 1.36±0.01
1.75±0.01
0.99947
0.99963
0.99948
0.99954
0.99951
136 17.41 0.030 - 0.400 1.432±0.003 1.254±0.006
0.99974
Table S1 lists the Avrami parameters for the full range of crystallization temperatures.
T c
= 134 o C was omitted from the data set of the 40 nm sample due to error in the
S4
measurement. For both bulk and confined samples the Avrami exponent, n , remained largely unaffected by the crystallization temperature. However, t
1/2
generally increased and k decreased with increasing crystallization temperature, both are generally observed trends.
Table S2. Values for 2θ of ( hkl ) reflections for the α-phase of bulk iPP, iPP-200, iPP-40 and iPP-15.
( h k l )
α
(110)
(040)
(130)
(111)
14.14
16.92
18.55
21.31
(131)/(041) 21.86
(150)/(060) 25.35
(200)
(220)
27.18
28.51
2θ ( o ) ( 1 ) d-spacing
(nm) 1
2θ ( o )
Bulk-iPP
0.626
0.524
0.478
0.417
0.406
0.351
0.328
0.313
14.19
17.06
18.67
21.14
21.88
25.66
---
28.35
2θ ( o ) iPP-200
14.01
16.78
18.30
---
---
---
26.95
---
2θ ( o ) iPP-40
14.19
16.90
18.54
21.13
21.98
---
27.00
---
2θ ( o ) iPP-15
13.98
16.81
18.59
---
---
---
26.86
---
S5
Recrystallization of iPP
Figure S3. Heating and cooling thermograms of as-purchased (bulk) and recrystallized iPP. Scan rate = 10 C min -1 . Second scan shown.
As-purchased iPP was recrystallized from p-xylene to remove any remaining catalyst and other impurities. The polymer was first brought to a boil in a ~ 1 wt% solution and heated for 10 min once the polymer had dissolved, then allowed to cool to room temperature. The resulting solution was filtered using Whatman Grade 1 filter paper and rinsed with methanol. The isolated powder was dried overnight under vacuum at 80 o C to remove residual solvent. Differential scanning calorimetry scans of the sample before and after recrystallization reveal stark differences, Figure S3. Firstly, the exothermic peak maximum shifted from ~ 123.5 o C to 120.6 o C following recrystallization. The shift was attributed to the removal of impurities from the as-purchased material. Secondly, a secondary peak appeared at ~ 147.5 o C of the endotherm, this peak may be due to low molecular weight chains melting.
S6
Experimental
Sample Preparation.
Isotactic polypropylene (iPP) was purchased from Sigma
Aldrich. The molecular weight and dispersity were determined to be M w
~ 243,000 g mol -
1 and 2.18, respectively using gel permeation chromatography. Following recrystallization from p-xylene to remove any remaining catalyst and impurities, the molecular weight and dispersity were 241,000 g mol -1 and 2.19, respectively. Resultant iPP powder was hot pressed into sheets at 166 o C with a load of 4 metric tons.
Anodic aluminum oxide (AAO) templates were fabricated from aluminum foil (99.999
%, Sigma Aldrich) using a two-step electrochemical oxidation method.
2 Anodization of aluminum in 0.3 M sulfuric acid solution at 25 V or in 0.3 M oxalic acid solution at 40 V resulted in self-ordered pores of 15 or 40 nm diameter, respectively.
An AAO template was placed directly on top of an iPP sheet, sandwiched between two glass slides, and fastened with binder clips. Infiltration of iPP into the AAO template was conducted under vacuum at 200 o C for 20-24 hours. The samples were then immediately quenched on a steel plate. Samples used for the X-day diffraction studies with cradle were annealed for an additional 30 minutes.
Scanning Electron Microscopy (SEM).
The top glass slide was detached and the excess bulk polymer was mechanically removed with sandpaper. The top, un-infiltrated layer of alumina was etched away using a 5 wt% sodium hydroxide solution. Then the intermediate layer of aluminum was removed using a solution of copper (II) chloride in hydrochloric acid at a concentration of 0.05 g ml -1 . The remaining alumina was then etched away using sodium hydroxide solution to release the nanostructures from the
S7
template. Released nanowires were imaged using a JEOL JSM-7500F scanning electron microscope.
Differential Scanning Calorimetry (DSC). The sample was placed in 5 wt% sodium hydroxide solution to remove the top alumina layer. A razor blade was then used to detach the sample from the bottom slide, and excess bulk polymer was removed using an
O
2
-plasma etcher (Harrick Plasma PDC-32G) and/or sandpaper. The etched surface was covered with Kapton tape and the sample was placed in a solution of copper (II) chloride and hydrochloric acid as before to remove the aluminum layer. Thermal analysis was performed using a TA Instruments Q200 differential scanning calorimeter. The instrument was calibrated using the Calibration Wizard software (TA Instruments
QSeries software). The heat flow (Tzero) calibration was performed using sapphire standards and the temperature (cell constant) was calibrated with indium at 10 o C min -1 .
All DSC measurements were performed under a nitrogen atmosphere using Tzero aluminum pans and lids. The sample weight ranged between ~ 5-15 mg, template plus polymer, depending on whether the sample was bulk or confined polymer.
X-Ray Diffraction (XRD).
The sample was removed from the bottom slide using a razor blade. Bulk polymer was removed from the template surface as described before.
θ/2θ diffraction patterns were collected in reflection mode (Bruker-AXS D8 Advance).
Copper K-α radiation was used with a wavelength of 0.154 nm. The instrument was operated using Commander Software and outfitted with a nickel filter. The scanning increment and integration time were 0.03 o and 1 s respectively. Then, the orientation of iPP infiltrated into AAO templates was investigated using a cradle. Tilt angle ( Ψ ) was varied from 0 o to 85 o in 1 o steps and with an integration time of 10 s. Data were
S8
collected by specifying the position of the incident and reflected beams, corresponding to a particular ( hkl ) plane, and collecting the intensities of the reflected rays as the sample stage was tilted by an angle Ψ . A schematic view of the experimental set-up for this analysis is shown below.
Figure S4.
A representation of a polymer-infiltrated AAO template investigated using
XRD. In this diagram, θ is fixed, and Ψ is allowed to vary.
Removal of the Bulk Surface Layer
Figure S5.
Heating (a) and cooling (b) thermograms of iPP infiltrated into an AAO template (200 nm pore diameter) exposed to varying durations of plasma treatment. Scan rate 10 o C min -1 . Second scan shown.
Figure S5 shows the effect of plasma etching on an AAO template (200 nm pore diameter) infiltrated with recrystallized iPP. The sample was exposed to 0, 1, or 2 hours of oxygen plasma, with the motivation of identifying the optimal etching duration.
S9
Without any etching, the untreated sample displayed a crystallization peak bearing a shoulder at slightly higher temperatures, Figure S5b. After one hour of plasma treatment the excess bulk layer was successfully removed, and the confined polymer was exposed.
This was confirmed by the appearance of a low-temperature crystallization peak in the cooling scan and the disappearance of the previously observed shoulder. After two hours of etching, the measured heat flow was greatly reduced, suggesting that the infiltrated polymer had been etched away. Therefore, one hour was selected as the optimal etching duration.
References.
1.
E. Clark, In Physical Properties of Polymers Handbook; Mark, J., Ed.; Springer
New York, 2007 , p 619-624.
2.
H. Masuda, K. Fukuda, Science 1995 , 268 , 1466-1468.
S10