Supplementary Materials for
advertisement

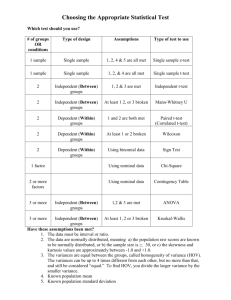
Supplementary Materials for MetaComp: A Comprehensive Analysis Tool for Comparative Metagenomics Peng Zhai, Jiangtao Guo, Xiaoqi Wang, Xiao Guo, Fumeng Wang, Fangmin Tian and Huaiqiu Zhu* State Key Lab for Turbulence and Complex Systems and Department of Biomedical Engineering, and Center for Quantitative Biology, Peking University, Beijing 100871, China *To whom correspondence should be addressed, Email: hqzhu@pku.edu.cn S1. Input data The input data of MetaComp can be represented as an Abundance Profile Matrix (APM). In order to obtain APM, users first have to align sequences (e.g. DNA, RNA or protein sequence) from metagenomic samples against a given database (e.g. Pfam, COG or GO). Then, total hit numbers of all sequences in one sample to each feature (e.g. Pfam ID or COG ID) is counted. The hit number of sample j to feature i is the value of 𝑐𝑖𝑗 (Table S1). In other words, 𝑐𝑖𝑗 is the total number of feature i observed in metagenomic sample j . Table S1. Input data of MetaComp. Feature Sample 1 Sample 2 … Sample n Feature 1 𝑐11 𝑐21 … 𝑐𝑛1 Feature 2 𝑐12 𝑐22 … 𝑐𝑛2 Feature 3 𝑐13 𝑐23 … 𝑐𝑛3 …… … … … … Feature m 𝑐1𝑚 𝑐2𝑚 … 𝑐𝑛𝑚 S2. Statistical test According to the type of input data, we have classified statistical tests into three modes: two samples test mode, multiple samples test mode and two sample groups test mode. (1) Two samples test mode As the sample size of Metagenome is generally large, we choose z-test instead of t-test as our recommendation for testing. Specifically, N1 and N2 are total counts of features in two samples. Then z-score for the feature Fi is: 𝑐 𝑐 1 1 𝑧𝑖 = (𝑁𝑖1 + 𝑁𝑖2 )⁄√𝑃(1 − 𝑃)(𝑁 + 𝑁 ) 𝑖1 𝑖2 𝑖1 𝑖2 (1) 𝑚 where 𝑁𝑖1 = ∑𝑚 𝑖=1 𝑐𝑖1 , 𝑁𝑖2 = ∑𝑖=1 𝑐𝑖2 , and 𝑃 = (𝑐𝑖1 + 𝑐𝑖2 )⁄(𝑁𝑖1 + 𝑁𝑖2 ). Since z-test is not valid, the prerequisite of z-test is min(𝑐𝑖1 , 𝑐𝑖2 ) ≤ 𝑧𝑖2 . (2) Multiple samples test mode Two samples tests between all conceivable pairs of samples are executed in this mode. For a specific feature i, the minimum of all conceivable p-values is considered as the p-value of this feature. Thus we can identify that this feature is significantly different in at least one pair of samples. (3) Two groups of samples test mode In MetaComp, we provide four statistical test methods (t-test, Paired t-test, Mann-Whitney U test and Wilcoxon sign-rank test) to assess whether a specific feature is significantly different between two groups. Users can choose a proper method themselves. Meanwhile, in order to facilitate the user without adequate statistic knowledge, MetaComp can automatically select the most suitable test method according to your input data. The appropriate statistical test method is chosen according to the contingency table shown in Table S2. Moreover, odds ratio test is implemented for evaluating the abundance of each feature. As the contingency table shown in Table S3, 𝑐𝑖𝑗 denotes the count of feature i in sample j. Considering the possibility of unevenness between two groups, an empirical continuity correction is introduced to improve the accuracy of the test (Sweeting, M.J. et al., 2004). Consequently, the statistics of the OR statistic for feature i is: log 2 𝑂𝑅(𝑖) = log 2 𝑅 1 )(𝑀22 + ) 𝑅+1 𝑅+1 1 𝑅 (𝑀12 + )(𝑀21 + ) 𝑅+1 𝑅+1 (𝑀11 + (2) where 𝑅 = 𝑀1 ⁄𝑀2 . According to the computation above, features can be classified as group 1 enriched (when log 2 𝑂𝑅(𝑖) > 1) and group 1 scarcity (when log 2 𝑂𝑅(𝑖) < 1). S3. Multiple Hypothesis Testing Correction A typical metagenomic profile consists of several hundreds to thousands of features (e.g. Pfam/Cog functional profiles). Therefore, direct application of statistical method described above may probably lead to large numbers of false positive. For example, choosing a threshold of 0.05 will result in 500 false positives in a profile contains 10000 features. Two correction methods are implemented in MetaComp to solve this problem, including default option false discovery rate (FDR) and an alternative option Bonferroni correction. Table S2. Method for comparison of two groups of samples independent Correlatedc Parametrica Non- parametricb t-test Paired t-test Mann-Whitney U test Wilcoxon sign-rank test a. Under the assumption that the variable follow a normal distribution b. Applicable when sample size is small and normality assumption is violated c. Consist of a sample of matched pairs of similar units, or one group of units that has been tested twice Table S3. Contingency table for odd ratio test Group 1(G1) Feature i 𝑀11 = ∑ Other Feature 𝑀21 = ∑ Total 𝑗∈𝐺1 𝑗∉𝐺1 Group 2(G2) 𝑐𝑖𝑗 𝑀12 = ∑ 𝑐𝑖𝑗 𝑀22 = ∑ 𝑀1 = 𝑀11 + 𝑀21 𝑗∈𝐺2 𝑗∉𝐺2 Total 𝑐𝑖𝑗 𝑛1 = 𝑀11 + 𝑀12 𝑐𝑖𝑗 𝑛2 = 𝑀21 + 𝑀22 𝑀2 = 𝑀12 + 𝑀22 S4. Environmental Factor Analysis This analysis is implemented by regression analysis via the lasso algorithm. MetaComp first normalizes the input data and environmental factors data before analysing. After that, the ith environmental factor in jth sample (Xij ) is considered as independent variable, and the jth frequency of a given feature (Yj ) is considered as dependent variable. The regression function is: m n Y j = ∑i αi Xji + ∑m≠n m,n βm,n X j × X j (3) where 𝑋𝑗𝑚 × 𝑋𝑗𝑛 means the co-effect of environmental factor 𝑋𝑗𝑚 and 𝑋𝑗𝑛 to feature Y𝑗 , 𝛼𝑖 , 𝛽𝑚,𝑛 represent the regression coefficients of the function. Moreover, the reliability of the regression coefficients is determined by p-value. Only when all p-values (p-values of all regression coefficients and p-value of the regression function) meet the prescribed standard, we shall accept the result of regression. Table S4. Part of the detailed result of the analysis of whale fall, Acid Mine Drainage, Sargasso Sea, and Minnesota soil metagenomic samples AMD Soil S.1a S.2 S.3 W.Boneb W.Mat W.Rib p-value q-value Function PF01036 0 1 344 354 332 0 0 0 1.60e-35 4.67e-34 Bacteriorhodopsin-like protein PF03814 99 17 3 4 7 0 0 1 5.22e-48 2.92e-46 Ion channel KdpA Potassium-transporting ATPase A subunit PF02705 0 87 15 30 30 10 0 1 2.27e-56 3.56e-54 APC K trans K+ potassium transporter PF01077 42 4870 62 51 71 57 45 37 0 0 NIR_SIR Nitrite and sulphite reductase 4Fe-4S domain a.S=Sargasso Sea b.W=whale fall Table S5. Part of the detailed result of relationship between metagenomic samples and environmental factors Feature DIPa COG0378 0 Oxygenb DIP&Oxygen p-value Correlation Annotation 0 3.303E-06 2.974E-02 9.006E-01 Ni2+-binding GTPase involved in regulation of expression and mature COG1921 1.157E-03 0 0 8.413E-02 8.223E-01 Selenocysteine synthase [seryl-tRNASer selenium transferase] COG0318 4.841E-03 0 0 8.972E-02 8.152E-01 Acyl-CoA synthetases (AMP-forming)/AMP-acid ligases II a.DIP=dissolved inorganic phosphate b.Oxygen=oxygen content References Sweeting, M.J. et al. (2004) What to add to nothing? Use and avoidance of continuity corrections in meta-analysis of sparse data, Statistics in medicine, 23(9), 1351-1375