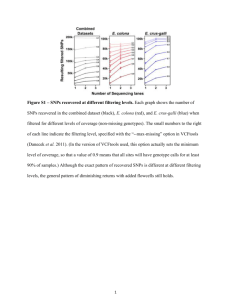
View/Open - Lirias
advertisement

Citation Van Geystelen A., Decorte R., Larmuseau M.H.D. (2013), Updating the Y-chromosomal phylogenetic tree for forensic applications based on whole genome SNPs Forensic Science International: Genetics, 7 (6), 573-580. Archived version Author manuscript: the content is identical to the content of the published paper, but without the final typesetting by the publisher Published version insert link to the published version of your paper http://www.sciencedirect.com/science/article/pii/S1872497313000896 Journal homepage insert link to the journal homepage of your http://www.fsigenetics.com/home. Author contact your email maarten.larmuseau@bio.kuleuven.be Klik hier als u tekst wilt invoeren. IR url in Lirias https://lirias.kuleuven.be/handle/123456789/ 395607 (article begins on next page) paper Abstract The Y-chromosomal phylogenetic tree has a wide variety of important forensic applications and therefore it needs to be state-of-the-art. Nevertheless, since the last 'official' published tree many publications reported additional Ychromosomal lineages and other phylogenetic topologies. Therefore, it is difficult for forensic scientists to interpret those reports and use an up-to-date tree and corresponding nomenclature in their daily work. Whole genome sequencing (WGS) data is useful to verify and optimize the current phylogenetic tree for haploid markers. The AMY-tree software is the first open access program which analyses WGS data for Y-chromosomal phylogenetic applications. Here, all published information is collected in a phylogenetic tree and the correctness of this tree is checked based on the first large analysis of 747 WGS samples with AMY-tree. The obtained result is one phylogenetic tree with all peer-reviewed reported Y-SNPs without the observed recurrent and ambiguous mutations. Nevertheless, the results showed that currently only the genomes of a limited set of Y-chromosomal (sub-)haplogroups is available and that many newly reported Y-SNPs based on WGS projects are false positives, even with high sequencing coverage methods. This study demonstrates the usefulness of AMY-tree in the process of checking the quality of the present Ychromosomal tree and it accentuates the difficulties to enlarge this tree based on only WGS methods. Keywords Haploid markers, Y-chromosome, phylogenetic tree, bio-informatics, Wholegenome SNP calling, Y-SNPs Introduction A state-of-the-art phylogeny of the human Y-chromosome based on bi-allelic polymorphisms is an essential tool for forensic genetics. Forensic scientists are taking advantage of the Y-chromosomal phylogenetic tree in their daily work, e.g. by checking the quality of datasets or by assigning geographical landscapes to specific lineages [1, 2]. Y-chromosomal single nucleotide polymorphisms (Y-SNPs) have a great capacity for detecting geographical origins as many lineages defined by Y-SNPs show a strong continent-specific [3, 4] and even intra-continent-specific distribution [5-7]. Their usefulness is illustrated by the fact that Y-SNP data are now also included in Y-chromosomal forensic databases, such as in the YHRD database [8]. Therefore, an up-to-date extended Y-chromosomal phylogeny based on these bi-allelic markers which are preferably unambiguous and non-recurrent but which have a high discrimination power is required for forensic applications. Since the publication of the latest 'official' Y-chromosomal phylogenetic tree by Karafet et al. [4], a continuous wave of new peer-reviewed articles which report changes to this tree are published. These changes include a new root and new basal clades [9, 10], modifications of the global backbone [3, 11], different phylogenetic topologies within a haplogroup [12-14], newly described subhaplogroups [15-17], or other phylogenetic positions for a certain mutation [18]. As these publications are not coordinated different names are given to the Y-chromosomal lineages for which the phylogenetic position is given in different topologies. Therefore, the currently overall reported Y-chromosomal tree is not clear and this makes it difficult for forensic researchers to use a uniform phylogenetic tree. Hence new initiatives to ensure more continuity in the report of the most recent phylogenetic Y-chromosomal tree are needed. Large whole genome sequencing (WGS) projects such as the 1000 Genomes project [19, 20] bring an opportunity to introduce the required uniformity in the reporting of the haploid Y-chromosomal tree. The analysis of whole Ychromosomes within male genomes allows verification and optimization of the currently used phylogenetic tree. WGS data has already proved to be useful in verifying and optimizing the phylogeny of the other haploid markers in the human genome i.e. the mitochondrial DNA (mtDNA). Relevant ambiguous markers and back-mutations which influence the interpretation of previous forensic and evolutionary genetic studies were detected based on these data [21]. Recently a new Y-chromosomal phylogenetic tree was built after a tabula rasa of the present Y-chromosomal tree by using only Y-SNPs from available WGS male samples [22]. By comparing this new phylogenetic tree with the currently used one, the backbone of the currently used phylogenetic tree was confirmed in this study. However, this new tree is not useful for forensic research because there is no link between currently used and newly reported lineages. Furthermore, the set of used genomes is not a good representation of all existing Y-chromosomal (sub-)haplogroups and geographical regions. There are also still too much false positive SNP calls in this WGS dataset. Alternatively, the AMY-tree software is the first open access program which academics and forensic professionals can use to verify and optimize the currently used Y-chromosomal tree by using WGS data [23]. The first AMYtree analysis was done based on 118 WGS samples and proved already its usefulness to verify and to optimize the present Y-chromosomal phylogenetic tree [23]. The aim of this study is to perform the largest reported screening of male genomes for Y-chromosomal phylogenetic applications based on the AMY-tree software. Firstly, we want to merge all newly Y-SNPs from recent peerreviewed publications since the latest 'official' Y-chromosomal phylogeny [4] into one single tree which is useful for forensic applications. Secondly, this updated Y-chromosomal phylogenetic tree needs to be checked for recurrent mutations, ambiguous SNPs and other difficulties for the (forensic) application of the tree. Thirdly, this study also wants to find out for which Y-chromosomal (sub-)haplogroups there is already WGS data available. Finally, investigating the possibilities to enlarge the Y-chromosomal phylogenetic tree based on the current Y-SNP detections in WGS data is the last aim of this study. Materials and methods Updated phylogenetic tree The latest updated phylogenetic tree of the Y-chromosome as it was published by Van Geystelen et al. [23] was manually updated based on recent descriptions of new Y-SNPs in academic research papers like Pamjav et al. [16] and Scozzari et al. [9]. As the exact phylogenetic position of a few new Y-SNPs was not given their position needed to be determined based on the results of AMY-tree of all WGS samples. Next, also recurrent mutations, ambiguous SNP-loci and wrongly defined mutation conversions within the newly updated Y-chromosomal tree were ascertained based on those AMY-tree results. WGS Y-SNPs dataset In order to check the manually updated phylogenetic tree and to optimise the AMY-tree software, a large dataset of whole genome Y-SNP calls was assembled. This dataset consists of 747 samples which represent 660 males, as several genomes were analysed in different projects. Within this dataset the genomes of eight males whose father's genome was also sequenced are present. The SNP calls were collected from four large WGS projects and several individual genome projects (Supplementary Materials Table S1). These projects differ from each other based on the used next-generation sequencing (NGS) platforms and sequence coverage. First, Complete Genomics made the SNP calls of 35 whole genomes of males available (http://www.completegenomics.com/public-data/69-Genomes/ 04 Jan 2013); those genomes were sequenced with a high sequencing coverage on the Complete Genomics Analysis (CGA) Platform [24]. Second, the Personal Genome Project (PGP) and Singapore Sequencing Malay Project (SSMP) also used this CGA platform. PGP is a project started to obtain and openly share human genome sequences in combination with health information. At the moment 40 male genomes were available (www.personalgenomes.org 04 Jan 2013). The SSMP on the other hand wanted to characterize the polymorphic variants in the population of Malays, an Austronesian group present in Southeast Asia and Oceania. Recently, the Y-SNP calls of 46 Malays were made publically available [25]. Next, the 1000 Genomes Project aims to provide a comprehensive resource on human genetic variation by sequencing more than 1000 human genomes. In 2010, SNP calls of 77 males were made available in the pilot phase [20] and two years later a set of 526 SNPs profiles were published as result of phase 1 of the Project [19]. As the 1000 Genomes project aims to sequence a large number of people, the sequencing coverage was lower than in the other projects. Finally, 23 additional samples were collected from several single genome projects [26-35, 36 and unpublished genomes of Guy Froyen]. AMY-tree modifications Several modifications to the AMY-tree software version 1.0 [23] were made for the assessment of the SNP calling quality in WGS data. This was necessary as the quality of the SNP calling influences the AMY-tree analysis of a sample and therefore also the interpretation of the result of the analysis [23]. The extra quality assessment is based on the results of the first AMY-tree run of a certain sample. This assessment assumes that the used phylogenetic tree is correct and that the assigned haplogroup after the first AMY-tree run is the actual haplogroup of that sample. The algorithm for the extra quality assessment is simple and comprehensible as shown in Figure 1. First, all Y-SNPs of the phylogenetic tree are selected except if the determined haplogroup of the first run is a paragroup (e.g. R1b1b2*). In the case of a paragroup, all Y-SNPs which are in sub-nodes of the main group of this paragroup are excluded from the selection. This is done in order to remove the influence of too much false positive SNP calls because when the haplogroup is not at the correct phylogenetic level, too much false positives and/or false negatives mutant Y-SNPs would be detected. Next, the expected state of all selected Y-SNPs is determined whereby all Y-SNPs in the path from the assigned sub-haplogroup till root are expected to be mutant and all others are expected to be ancestral. The state of each Y-SNP is also determined in the reference genome. Thereafter, these expectations of Y-SNP state are converted into expectations of ‘called’ or ‘not called’ in the next step based on the expected state and the state in the reference genome. Consider a Y-SNP in the path from sub-haplogroup till root, if the status of that SNP in the reference sequence is mutant, the Y-SNP is expected to be 'not called' else the expectation is 'called'. Consider a Y-SNP not in the path from sub-haplogroup till root, if the status of that Y-SNP in the reference sequence is mutant, the YSNP is expected to be 'called' else the expectation is 'not called'. Then, these expectations are compared to the state in the sample such that the number of true positive, false positive, true negative and false negative SNP calls will be determined. At last, the quality of a sample is expressed in several measures of quality: Matthews correlation coefficient (MCC), accuracy, sensitivity, specificity, precision, recall and F1-score (Supplementary method). When the MCC is larger or equal to 0.95 the SNP calling quality is called excellent, otherwise it is called low. The quality will be given in the output file of AMYtree. When the SNP calling quality is low, caution has to be taken about the result of the AMY-tree analysis due to the high occurrence of false negative and false positive SNPs. When the quality is excellent, the results of AMY-tree are considered to be valuable for the control of the currently used phylogenetic tree of the Y-chromosome and for the increase of its resolution. As such, a better YSNP call quality assessment is implemented in AMY-tree version 1.1 compared to the earlier version [23]. Next, when a sample was run in AMY-tree in the 'sufficient' mode such that the reference genome was taken into account but the determined haplogroup belongs to R-M269 and the MCC lower than 0.95 a second AMY-tree run needs to be executed but in the 'insufficient' mode. This is important as a MCC lower than 0.95 indicates that this result of the first run is too much influenced by the reference genome. Finally, another small modification to AMY-tree is based on the fact that Z381, L2 as well as L20 are mutant in the reference genome and although AMY-tree version 1.0 already had a quite complex system to filter out the influence of the reference genome even on samples belonging to haplogroup R, it was not yet efficient enough. Therefore, when both Z381 and L2 or both Z381 and L20 are mutant in the first AMY-tree run, e.g. the ancestral SNP was not called, the sample will be handled as insufficient in a second AMY-tree run, such that the Y-SNPs of the reference genome are not used anymore when determining the haplogroup. By including these modifications even more certainty is build in AMY-tree version 1.1 in comparison with the previous version [23]: for samples belonging to a R-M269 sub-haplogroup the reference genome is only taken into account when the SNP calling quality is excellent after the first run in the ‘sufficient’ mode. The cut-offs to assess the Y-SNP calling quality is optimised based on all 747 genomes by performing several test runs and manual analyses of the genomes and by checking it with the results in Van Geystelen et al. [23] and with the publications of the genomes whereby a Y-chromosomal analysis was already performed. Y-SNP detecting The Y-SNPs which are present in the WGS samples but which are not yet included in the updated phylogenetic tree were detected by AMY-tree. Only those Y-SNPs from WGS samples with an excellent Y-SNP calling quality were used. But that was not the only constraint for the potentially relevant YSNPs; they must also be positioned in the unique regions of the Y-chromosome as read mapping and variant detection difficulties are expected due to the high frequency of repeated sequences on the Y-chromosome. So, Y-SNPs in the pseudoautosomal, heterochromatic, X-transposed and ampliconic segments [37] of the male-specific part of the genome as reported by Wei et al. [22] were excluded. Results In total 747 samples are analysed by the updated version of the AMY-tree software with the updated phylogenetic Y-chromosomal tree. There are 131 samples of 126 individuals with an excellent Y-SNP calling quality, i.e. MCC ≥ 0.95, which are mostly obtained from Complete Genomics and the Personal Genome Project. The remaining 616 samples have a low calling quality and are mostly obtained from the 1000 Genomes Project pilot and phase 1 (Table S1). Updated tree for forensic applications The state-of-the-art Y-chromosomal tree is manually updated based on all published Y-SNPs from academic studies. After the AMY-tree runs of all 747 samples, all Y-SNPs of which no exact phylogenetic position was given in the publications could now be included in the updated phylogenetic tree, for example there were two new Y-SNPs reported in [16] within sub-haplogroup R-M198 without clear phylogenetic positions (Figure 2). The results with an excellent SNP calling quality also showed several recurrent Y-SNPs in the phylogenetic tree which cause the determination of multiple haplogroups for some samples. After ruling out that the recurrent SNPs are sample- or project-specific, these SNPs are removed from the phylogenetic tree; an overview of the three observed recurrent SNPs of which enough evidence was available, is given in Table S2. These modifications led to the final updated tree version 1.1 which includes 359 Y-chromosomal lineages and 721 Y-SNP markers. The final tree and its corresponding mutation conversions for all the Y-SNPs in the tree can be found in Tables S3 and S4. Sub-haplogroup determining The determined haplogroups of all 747 samples obtained by the AMY-tree analysis based on the final updated tree version 1.1 is given in Table S1. Only for 14 samples no haplogroup can be determined and for three other samples multiple haplogroups are determined by AMY-tree. The distribution of the MCC of the 730 samples for which an unambiguous haplogroup is determined is given in Figure 3. Only a minority of the samples has an excellent SNP calling quality with a MCC ≥ 0.95; a MCC lower than 0.95 means that less than 97.5% of the negative and positive predictions are correct. Overall, 17 different haplogroups and 106 sub-haplogroups are present in the dataset. When considering only the samples of excellent quality 10 different haplogroups and 47 sub-haplogroups remain. Figure 4 and Table S5 give an overview of those (sub-)haplogroups and their frequencies. The samples of paternally related samples present in the dataset are of particular interest because they are considered to represent the same Y-chromosome. All the samples of one family with eight members sequenced by Complete Genomics are determined to belong to R-P312*. The paternal grandfather of that family is also analysed in the 1000 Genomes project and there he is determined as P* [P-92R7*]. The first attempt of AMY-tree to determine the sub-haplogroup of that sample of 1000 Genomes led to the sub-haplogroup R-L2 which has a higher phylogenetic level than the haplogroups of the Complete Genomics samples. However, as the MCC value of that sample is smaller than 0.95 the sub-haplogroup determination is done again but without the influence of the reference genome. This led to the final sub-haplogroup P92R7* which has a less accurate phylogenetic level than that of Complete Genomics. Thus, the influence of the reference genome can sometimes cause a too high or too low phylogenetic level when the new modifications which were made to AMY-tree version 1.1 would not have been applied. Detecting new Y-SNPs The large amount of available samples leads to a huge amount of newly reported Y-SNPs, i.e. Y-SNPs that are not yet present in the updated phylogenetic tree version 1.1. In total 108,681 new Y-SNPs are reported in all 660 male genomes; when an individual is analysed in more than one project only the sample with the highest MCC value is used. The majority of the SNPs appears in only a few samples: 62% appears in only one sample and 16% is present in two samples as shown in Figure 5A. In the 126 male genomes with an excellent Y-SNP calling quality 50,430 new Y-SNPs are reported. These SNPs also come with a high frequency of low occurrences in the excellent genomes: 57% is unique and 11% appears in two samples. When only the regions within the Y-chromosome which are identified as unique are taken into account, a much lower number of new Y-SNPs is detected. In total 35,503 new Y-SNPs are reported in the 660 male genomes and 15,208 new Y-SNPs in the genomes with excellent Y-SNP calling quality. The same patterns of occurrence for these Y-SNPs are observed as with the non-filtered Y-SNPs: the majority of the new reported Y-SNPs appeared in only one or two samples of the WGS dataset as shown in Figure 5B. The genomes of eight males whose biological father's genome is also sequenced are present in the dataset: one family of eight males including the father, the son and the six grandsons next to one father-son pair. In the family of eight paternal relatives 5,155 new SNPs are reported on the whole Y-chromosome and a large number of these SNPs is found in only one of the eight individuals as shown in Figure 6A. The number of Y-SNPs decreases every time the number of samples in which the Y-SNP occurs increases except for the occurrence in all-exceptone and all samples of the family. When all Y-SNPs which also occur in any other sample with excellent SNP calling quality are removed only less than 20% of the Y-SNPs remains but the distribution of the number of Y-SNPs per occurrence stays the same as the black bars in Figure 6A show. The same comparison is made when only the SNPs in the unique part of the Ychromosome are selected: the same pattern as with all SNPs is visible in Figure 6B. However, for each number of occurrences the number of truly unique SNPs, i.e. SNPs that do not occur in other genomes outside the family, is much higher. Within the father-son pair 2,181 new SNPs are reported on the whole Y- chromosome for which the difference between occurrence in only one and both genomes is relatively small, also for the proportion of SNPs which do not occur in the other genomes with an excellent Y-SNP quality (Figure 6A). Remarkably, there are more new SNPs present in both samples than in one sample when the Y-SNPs which are not located in the unique region of the Ychromosome are removed (Figure 6B). The effect of the increasing number of unique Y-SNPs that do not occur in other genomes as seen with the previous family is also in the father-son pair present. Discussion The present study realises the first large screening of male genomes for phylogenetic applications of the Y-chromosome. Based on this screening of 747 male samples an update has been made of the AMY-tree software and of a state-of-the-art Y-chromosomal phylogenetic tree was established for forensic scientists. Furthermore, also recommendations for future sequencing projects dealing with a broader selection panel of Y-chromosomal haplogroup samples and for the validation of newly detected Y-SNPs are made. First, the large screening of the 747 male Y-chromosome samples revealed that the SNP calling quality of a few samples was overestimated in AMY-tree version 1.0. As these samples belong to sub-haplogroup R-M269 they are very similar to the reference genome which is used to estimate the SNP calling quality of a sample [23]. To remove this SNP calling quality overestimation the influence of the reference genome is excluded for all samples belonging RM269 with a low SNP call quality. That is why several modifications are made and implemented in AMY-tree version 1.1. Second, an update of the currently used Y-chromosomal phylogenetic tree was realised based on the large database of 747 available samples. At the moment this tree is the most state-of-the-art tree applicable for forensic geneticists: all Y-SNPs which are reported in academic publications till today are included and all ambiguous markers are excluded to avoid wrong Y-SNP interpretations (Table S3). As often the case in the literature the phylogenetic relationship between new Y-SNPs and earlier reported Y-SNPs in the same (sub)haplogroup are not given. By using the AMY-tree results it is possible to find out the concrete phylogenetic level of each Y-SNP relative to the other already known SNPs. For example, Pamjav et al. [16] described and genotyped two new Y-SNPs Z93 and Z280 within R-M198, although the exact phylogenetic positions in relationship with the other lineages within R-M198 were not given. The presence of both Z93 and Z80 is checked in all samples belonging to the sub-haplogroup R-M198, Z93 occurred in three R-M198* samples and in none of the other samples. Also Z280 does not occur in any sample except in one RM198* sample. Therefore, both Z93 and Z280 are placed in the phylogenetic tree as sub-haplogroups of R-M198 as shown in Figure 2. The choice is made for a phylogenetic tree in table format as described by Van Geystelen et al. [23] instead of a branching diagram because the tree is very large and will become larger in the future. Therefore, the table format can be adapted more easily than the diagram and it is also more manageable. Not only newly reported Y-SNPs are included in the new updated tree but also ambiguous Y-SNPs are excluded as they can complicate the Y-chromosomal applications for forensic studies. As previously described [38], the most relevant ambiguous Y-SNPs are recurrent SNPs which have a paralogous distribution along the phylogenetic tree and which have thus mutant alleles in at least two independent Y-chromosomal lineages. Based on the screening of the 747 analysed samples, three Y-SNPs are recognized for the first time as recurrent (Table S2). There are no other indications for recurrent mutations based on the present WGS dataset. Therefore we may assume, in most cases, that males which both have the mutant allele for a Y-SNP used in the updated tree (Table S3) have received this mutant allele from one common ancestor and not by convergent evolution. Next, also all Y-SNPs which could not be analysed by WGS for one reason or another are excluded from the updated tree, although it may be possible to genotype these SNPs correctly with Sanger sequencing methods. For example, all hundred E-M2 samples in the dataset did not reveal the mutant allele for Y-SNP V95 as expected by earlier publications [14, 39]. The reason for this remarkable result may be that V95 is not well validated but more likely it is the result of a bad SNP calling in the Ychromosomal region around V95 by the current WGS methods. For some YSNPs also a mutation conversion is found which is different from the reported one. For example, another ancestral and mutant allele are observed for Y-SNP M426 than reported by Rootsi et al. [40]. Since the Y-chromosome has a very complex origin it also has a lot of non-unique regions which complicate the analysis of WGS data [37]. The current reference genome GRCh37 shows the evolution of the Y-chromosome and its numerous resulting non-unique regions. So the reason for this wrong analysis of M426 may be the position of this SNP in one of the non-unique regions of the Y-chromosome as defined earlier [22, 37]. To have an unambiguous phylogenetic tree, we excluded all the Y-SNPs for which no reliable signal with the WGS methods could be found. In the end an updated Y-chromosomal tree which includes 359 Y-chromosomal lineages and more than 721 Y-SNP markers (Table S3) is obtained and this tree is the basic tool to develop and optimize Y-SNP-multiplexes for forensic applications [41, 42]. Third, the distribution of the analysed haplogroups per ancestral continent in the current dataset corresponds with the known distributions [3, 4]. Nevertheless, there is not yet a representative set of all phylogenetic sub-haplogroups available. In total 17 haplogroups and 106 sub-haplogroups are reported in the analysis of the whole dataset. However, the SNP calling quality is low (MCC < 0.95) for most of the analysed samples as most data are obtained from the 1000 Genomes project and therefore the sub-haplogroup determination of these data is not completely reliable. When considering only the samples of excellent quality (MCC ≥ 0.95) 10 haplogroups and 47 sub-haplogroups remain and this corresponds with only 13% of the total number lineages described so far. Most of the Y-chromosomes are assigned to haplogroups E, O and R. Therefore, when the set of WGS Y-chromosomes will be enlarged the current phylogenetic tree as well as the analysis of Wei et al. [22] to calibrate the tree can be optimized. Finally, the screening of the dataset also revealed that new in silico and in vitro methods are required to verify new Y-SNPs based on WGS methods. As earlier mentioned [23], a huge number of new Y-SNPs are false positives when the genomes were sequenced with low coverage and consequently the called SNPs will have a low quality. These false positive SNP calls disturb the determination of the correct (sub-)haplogroup of the sample; consequently the AMY-tree software has to correct for them by applying several additional methods in the analysis [23]. The high number of false positive Y-SNPs - even detected in WGS samples with an excellent SNP calling quality - is observed by comparing genomes of paternal relatives. Within the eight paternally related samples belonging to one family, which are genotyped by Complete Genomics, 949 newly reported Y-SNPs in the full Y-chromosome and 88 ones in the unique regions of the Y-chromosome are found in at least one but not all family members (Figure 6). Despite the high coverage of these genomes and the excellent SNP calling quality this is still a very high number of newly reported Y-SNPs which are most likely to be false based on the mutation rate on the Y- chromosomes calculated based on a deep-rooting pedigree [43] and based on human-chimpanzee comparisons [44]. A similar conclusion can be made based on the father-son pair in the dataset which is also sequenced with a high coverage by Complete Genomics (Figure 6). The Y-SNP results show that adding new lineages to the Y-chromosomal phylogenetic tree only based on WGS is not evident. Therefore, making tabula rasa and building a new tree based on all WGS Y-chromosomes as done by Wei et al. [22] is not an option when the tree is going to be used in forensics. Each new Y-SNP and consequently each Y-chromosomal lineage has to be validated independently from the WGS data before adding them to the updated phylogenetic tree. The validation status of many potential polymorphic Y-SNPs (> 1% in population) is often unclear but this can be resolved by the sharing of genomic data among genetic genealogists which are interested in finding new Y-SNPs to resolve their particular paternal ancestry [45]. Therefore, this is an area in which closer collaborations between amateurs and forensic academics could prove to be particularly useful [13]. Nevertheless, it is required that new in silico methods will be designed to select good and relevant candidates of YSNPs for the validation. For example, an interesting criterion is the position of the Y-SNPs: it is more interesting to validate only the SNPs located in the unique regions of the Y-chromosome as there are many non-unique regions due to the evolutionary history of this chromosome [37]. The lower number of false positive SNP calls in these unique regions in comparison with those in the full Y-chromosome is clearly demonstrated in the father-son pair. The number of new Y-SNPs reported in only one of the two samples is higher than the new ones reported in both samples based on the whole Y-chromosome (Figure 6A) in contrast with the situation based on the unique regions of the Y-chromosome (Figure 6B). Conclusions Based on the largest screening of male genomes with 747 samples in total, the most up-to-date Y-chromosomal phylogenetic tree for forensic applications is compiled. Future publications which will report new Y-SNPs have to situate their phylogenetic positions in this tree to guarantee the continuity between old and new publications. At this moment, forensic scientists as well as evolutionary biologists and genetic genealogists are lost in the many reports of newly described Y-chromosomal lineages [46]. Therefore, initiatives as AMYtree which optimize the phylogeny based on peer-reviewed publications are required [23]. This is already the case for the mitochondrial genome with the Phylotree initiative of van Oven and Kayser [47]. Nevertheless, to optimize the current updated phylogenetic tree for the human Y-chromosome more high quality genomes of a broader set of (sub-)haplogroups than the frequent haplogroups E, O and R are required. Also a higher effort in the validation of reported Y-SNPs by new in silico and in vitro methods is required. Acknowledgements The authors want to thank Tom Wenseleers, Manfred Kayser, Jean-Jacques Cassiman, Tom Havenith, Hendrik Larmuseau and Lucrece Lernout for useful discussions and comments. Thanks also to Guy Froyen (VIB, KU Leuven), Richard Rocca (independent researcher), Cuiping Pan (Stanford University) and Andreas Keller (Saarland University) for providing us yet unpublished and published called SNPs of whole genome sequencing projects. Maarten H.D. Larmuseau is postdoctoral fellow of the FWO-Vlaanderen (Research Foundation Flanders). This study was funded by the KU Leuven BOF Centre of Excellence Financing on 'Eco and socio evolutionary dynamics' (Project number PF/2010/07) and on 'Centre for Archaeological Sciences 2 (CAS 2) New methods for research in demography and interregional exchange'. Authors’ contributions Research design & supervision: MHDL; Programming: AVG; Writing: MHDL & AVG; Commenting on manuscript: AVG, RD & MHDL. Conflict of interest The authors declare no conflict of interest. References [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] M. Kayser, Y-chromosomal markers in forensic genetics, in: R. Rapley and W. D., Editors, Molecular Forensics, John Wiley & Sons Ltd: Chichesters. 2007, pp. 141-161. J.M. Butler, Chapter 13 Y-Chromosomal DNA Testing, in: J.M. Butler, Editor, Advanced Topics in Forensic DNA Typing: Methodology, Academic Press: London. 2011, pp. 371-403. J. Chiaroni, P.A. Underhill, and L.L. Cavalli-Sforza, Y chromosome diversity, human expansion, drift, and cultural evolution, Proceedings of the National Academy of Sciences of the United States of America. 106 (2009) 20174-20179. T.M. Karafet, F.L. Mendez, M.B. Meilerman, P.A. Underhill, S.L. Zegura, and M.F. Hammer, New binary polymorphisms reshape and increase resolution of the human Y chromosomal haplogroup tree, Genome Research. 18 (2008) 830-838. M.H.D. Larmuseau, N. Vanderheyden, M. Jacobs, M. Coomans, L. Larno, and R. Decorte, Micro-geographic distribution of Y-chromosomal variation in the central-western European region Brabant, Forensic Science International-Genetics. 5 (2011) 95-99. F. Cruciani, B. Trombetta, C. Antonelli, R. Pascone, G. Valesini, V. Scalzi, G. Vona, B. Melegh, B. Zagradisnik, G. Assum, et al., Strong intra- and inter-continental differentiation revealed by Y chromosome SNPs M269, U106 and U152, Forensic Science International-Genetics. 5 (2011) E49-E52. M.H.D. Larmuseau, J. Vanoverbeke, G. Gielis, N. Vanderheyden, H.F.M. Larmuseau, and R. Decorte, In the name of the migrant father - Analysis of surname origin identifies historic admixture events undetectable from genealogical records., Heredity. 109 (2012) 90-95. S. Willuweit and L. Roewer, Y chromosome haplotype reference database (YHRD): Update, Forensic Science International-Genetics. 1 (2007) 83-87. R. Scozzari, A. Massaia, E. D'Atanasio, N.M. Myres, U.A. Perego, B. Trombetta, and F. Cruciani, Molecular dissection of the basal clades in the human Y chromosome phylogenetic tree, Plos One. 7 (2012) e49170. F. Cruciani, B. Trombetta, A. Massaia, G. Destro-Bisol, D. Sellitto, and R. Scozzari, A revised root for the human Y chromosomal phylogenetic tree: The origin of patrilineal diversity in Africa, American Journal of Human Genetics. 88 (2011) 814-818. S. Fornarino, M. Pala, V. Battaglia, R. Maranta, A. Achilli, G. Modiano, A. Torroni, O. Semino, and S.A. Santachiara-Benerecetti, Mitochondrial and Y-chromosome diversity of the Tharus (Nepal): a reservoir of genetic variation, Bmc Evolutionary Biology. 9 (2009) 154. F.L. Mendez, T.M. Karafet, T. Krahn, H. Ostrer, H. Soodyall, and M.F. Hammer, Increased resolution of Y chromosome haplogroup T defines relationships among populations of the Near East, Europe, and Africa, Human Biology. 83 (2011) 39-53. L.M. Sims, D. Garvey, and J. Ballantyne, Improved resolution haplogroup G phylogeny in the Y-chromosome, revealed by a set of newly characterized SNPs, Plos One. 4 (2009) e5792. B. Trombetta, F. Cruciani, D. Sellitto, and R. Scozzari, A new topology of the human Y chromosome haplogroup E1b1 (E-P2) revealed through the use of newly characterized binary polymorphisms, PLoS One. 6 (2011) e16073. M.S. Jota, D.R. Lacerda, J.R. Sandoval, P.P.R. Vieira, S.S. Santos-Lopes, R. Bisso-Machado, V.R. Paixao-Cortes, S. Revollo, C. Paz-Y-Mino, R. Fujita, et al., A new subhaplogroup of native American Y-chromosomes from the Andes, American Journal of Physical Anthropology. 146 (2011) 553-559. H. Pamjav, T. Feher, E. Nemeth, and Z. Padar, Brief communication: New Y-chromosome binary markers improve phylogenetic resolution within haplogroup R1a1, American Journal of Physical Anthropology. 149 (2012) 611-615. [17] [18] [19] [20] [21] [22] [23] [24] [25] [26] [27] [28] [29] [30] [31] [32] [33] [34] [35] [36] [37] [38] [39] [40] N.M. Myres, S. Rootsi, A.A. Lin, M. Jarve, R.J. King, I. Kutuev, V.M. Cabrera, E.K. Khusnutdinova, A. Pshenichnov, B. Yunusbayev, et al., A major Y-chromosome haplogroup R1b Holocene era founder effect in Central and Western Europe, European Journal of Human Genetics. 19 (2011) 95-101. S. Yan, C.C. Wang, H. Li, S.L. Li, L. Jin, and G. Consortium, An updated tree of Y-chromosome Haplogroup O and revised phylogenetic positions of mutations P164 and PK4, European Journal of Human Genetics. 19 (2011) 1013-1015. T. 1000 Genomes Project Consortium, An integrated map of genetic variation from 1,092 human genomes, Nature. 491 (2012) 56-65. D.L. Altshuler, R.M. Durbin, G.R. Abecasis, D.R. Bentley, A. Chakravarti, A.G. Clark, F.S. Collins, F.M. De la Vega, P. Donnelly, M. Egholm, et al., A map of human genome variation from population-scale sequencing, Nature. 467 (2010) 1061-1073. A.T. Duggan and M. Stoneking, A highly unstable recent mutation in human mtDNA, American Journal of Human Genetics. 9 (2013) 279-284. W. Wei, Q. Ayub, Y. Chen, S. McCarthy, Y. Hou, I. Carbone, Y. Xue, and C. Tyler-Smith, A calibrated human Y-chromosomal phylogeny based on resequencing, Genome Research. 23 (2013) 388-395. A. Van Geystelen, R. Decorte, and M.H.D. Larmuseau, AMY-tree: an algorithm to use whole genome SNP calling for Y chromosomal phylogenetic applications, BMC Genomics. 14 (2013) 101. R. Drmanac, A.B. Sparks, M.J. Callow, A.L. Halpern, N.L. Burns, B.G. Kermani, P. Carnevali, I. Nazarenko, G.B. Nilsen, G. Yeung, et al., Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays, Science. 327 (2010) 78-81. L.-P. Wong, R.T.-H. Ong, W.-T. Poh, X. Liu, P. Chen, R.Q. Li, K.K.-Y. Lam, N.E. Pillai, K.-S. Sim, H. Xu, et al., Deep whole-genome sequencing of 100 Southeast Asian Malays, American Journal of Human Genetics. 92 (2013) 1-15. S.C. Schuster, W. Miller, A. Ratan, L.P. Tomsho, B. Giardine, L.R. Kasson, R.S. Harris, D.C. Petersen, F.Q. Zhao, J. Qi, et al., Complete Khoisan and Bantu genomes from southern Africa, Nature. 463 (2010) 943-947. P. Tong, J.G.D. Prendergast, A.J. Lohan, S.M. Farrington, S. Cronin, N. Friel, D.G. Bradley, O. Hardiman, A. Evans, J.F. Wilson, et al., Sequencing and analysis of an Irish human genome, Genome Biology. 11 (2010) R91. A. Keller, A. Graefen, M. Ball, M. Matzas, V. Boisguerin, F. Maixner, P. Leidinger, C. Backes, R. Khairat, M. Forster, et al., New insights into the Tyrolean Iceman's origin and phenotype as inferred by whole-genome sequencing, Nature communications. 3 (2012) 698. R. Chen, G.I. Mias, J. Li-Pook-Than, L.H. Jiang, H.Y.K. Lam, R. Chen, E. Miriami, K.J. Karczewski, M. Hariharan, F.E. Dewey, et al., Personal omics profiling reveals dynamic molecular and medical phenotypes, Cell. 148 (2012) 1293-1307. J.M. Rothberg, W. Hinz, T.M. Rearick, J. Schultz, W. Mileski, M. Davey, J.H. Leamon, K. Johnson, M.J. Milgrew, M. Edwards, et al., An integrated semiconductor device enabling nonoptical genome sequencing, Nature. 475 (2011) 348-352. D. Pushkarev, N.F. Neff, and S.R. Quake, Single-molecule sequencing of an individual human genome, Nature Biotechnology. 27 (2009) 847-850. M. Rasmussen, Y.R. Li, S. Lindgreen, J.S. Pedersen, A. Albrechtsen, I. Moltke, M. Metspalu, E. Metspalu, T. Kivisild, R. Gupta, et al., Ancient human genome sequence of an extinct PalaeoEskimo, Nature. 463 (2010) 757-762. S.M. Ahn, T.H. Kim, S. Lee, D. Kim, H. Ghang, D.S. Kim, B.C. Kim, S.Y. Kim, W.Y. Kim, C. Kim, et al., The first Korean genome sequence and analysis: Full genome sequencing for a socioethnic group, Genome Research. 19 (2009) 1622-1629. D.A. Wheeler, M. Srinivasan, M. Egholm, Y. Shen, L. Chen, A. McGuire, W. He, Y.J. Chen, V. Makhijani, G.T. Roth, et al., The complete genome of an individual by massively parallel DNA sequencing, Nature. 452 (2008) 872-U5. J. Wang, W. Wang, R.Q. Li, Y.R. Li, G. Tian, L. Goodman, W. Fan, J.Q. Zhang, J. Li, J.B. Zhang, et al., The diploid genome sequence of an Asian individual, Nature. 456 (2008) 60-U1. B.A. Peters, B.G. Kermani, A.B. Sparks, O. Alferov, P. Hong, A. Alexeev, Y. Jiang, F. Dahl, Y.T. Tang, J. Haas, et al., Accurate whole-genome sequencing and haplotyping from 10 to 20 human cells, Nature. 487 (2012) 190-195. H. Skaletsky, T. Kuroda-Kawaguchi, P.J. Minx, H.S. Cordum, L. Hillier, L.G. Brown, S. Repping, T. Pyntikova, J. Ali, T. Bieri, et al., The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes, Nature. 423 (2003) 825-U2. S.M. Adams, T.E. King, E. Bosch, and M.A. Jobling, The case of the unreliable SNP: recurrent back-mutation of Y-chromosomal markers P25 through gene conversion, Forensic Science International. 159 (2006) 14-20. B. Trombetta, F. Cruciani, P.A. Underhill, D. Sellitto, and R. Scozzari, Footprints of X-to-Y gene conversion in recent human evolution, Molecular Biology and Evolution. 27 (2010) 714-725. S. Rootsi, N.M. Myres, A.A. Lin, M. Järve, R.J. King, I. Kutuev, V.M. Cabrera, E.K. Khusnutdinova, K. Varendi, H. Sahakyan, et al., Distinguishing the co-ancestries of [41] [42] [43] [44] [45] [46] [47] haplogroup G Y-chromosomes in the populations of Europe and the Caucasus, European Journal of Human Genetics. 20 (2012) 1275-1282. S. Caratti, S. Gino, C. Torre, and C. Robino, Subtyping of Y-chromosomal haplogroup E-M78 (E1b1b1a) by SNP assay and its forensic application, International Journal of Legal Medicine. 123 (2009) 357-360. C. Bouakaze, C. Keyser, S. Amory, E. Crubézy, and B. Ludes, First successful assay of Y-SNP typing by SNaPshot minisequencing on ancient DNA, International Journal of Legal Medicine. 121 (2007) 493-499. Y.L. Xue, Q.J. Wang, Q. Long, B.L. Ng, H. Swerdlow, J. Burton, C. Skuce, R. Taylor, Z. Abdellah, Y.L. Zhao, et al., Human Y chromosome base-substitution mutation rate measured by direct sequencing in a deep-rooting pedigree, Current Biology. 19 (2009) 1453-1457. Y. Kuroki, A. Toyoda, H. Noguchi, T.D. Taylor, T. Itoh, D.S. Kim, D.W. Kim, S.H. Choi, I.C. Kim, and H.H. Choi, Comparative analysis of chimpanzee and human Y chromosomes unveils complex evolutionary pathway, Nature Genetics. 38 (2006) 158-167. T.E. King and M.A. Jobling, What's in a name? Y chromosomes, surnames and the genetic genealogy revolution, Trends in Genetics. 25 (2009) 351-360. M.H.D. Larmuseau, A. Van Geystelen, M. van Oven, and R. Decorte, Genetic genealogy comes of age - Perspectives on the use of deep-rooted pedigrees in human population genetics., American Journal of Physical Anthropology. (In press). M. van Oven and M. Kayser, Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation, Human Mutation. 30 (2008) E386-E394. Figures Select Y-SNPs based on determined haplogroup Expected state ancestral/mutant State in reference genome Expected situation in sample Actual situation in sample called/not called called/not called TP, FP, TN, FN Measures Figure 1. Workflow of the quality assessment algorithm of the AMY-tree, version 1.1. First, certain Y-SNPs of the Y-chromosomal phylogenetic tree are selected based on the determined haplogroup of the first run in order to avoid too much false positive Y-SNPs. Next, the expected state (ancestral or mutant) of the selected SNPs is determined based on the haplogroup and the phylogenetic tree. These expectations of state are converted to expectations of called or not called based on the SNP state in the reference genome. These expectations are then compared to the actually called SNPs of the sample such that the number of true positives (TP), false positives (FP), true negatives (TN) and false negatives (FN) will be determined. Finally, several different measures will be calculated. M198, M417, M512, M514, M515, Page7 R-M198* R-M56 M157.1 R-M157.1 M87, M204 R-M87 P98 R-P98 PK5 R-PK5 M434 R-M434 M458 R-M458* M334 R-M334 Page68 R-Page68 Z280 R-Z280 Z93 R-Z93 M56 Figure 2. Overview of the position of the newly added sub-haplogroups RZ280 and R-Z93 (given in bold) within R-M198. Number of samples frequency 160 80 0 0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 Matthew's Correlation Matthew’s correlationCoefficient coefficient Figure 3. Distribution of the Matthews correlation coefficient of the 730 samples for which an unambiguous haplogroup could be determined. 0.9 1.0 Number of available WGS genomes 165 All genomes 110 55 Good quality genomes 0 A B C D E F G H I J K L M N O P Q R Haplogroups Figure 4. Frequency of whole genome sequencing (WGS) genomes per haplogroup in the dataset of all samples (black) and in the dataset of samples with an excellent quality, i.e. MCC ≥ 0.95 (grey). S T 70000 A 35000 Number of Y-SNPs Whole Y-chromosome 70000 All genomes 0 1 2 35000 3 4 5 6 Excellent quality genomes 7 8 9 10 more 9 10 more Number of occurrences of Y-SNP 0 1 B 2 3 4 5 6 7 8 Unique regions Y-chromosome 24000 Number of occurrences of Y-SNP 12000 24000 0 12000 1 2 3 4 5 6 7 8 9 10 more Number of occurrences of Y-SNP Figure 5. Number of new Y-SNPs per number of occurrence in the full WGS 0 dataset: (A) SNPs 1in the2 whole Y-chromosome and7(B) SNPs in only the 3 4 5 6 8 9 10 more identified unique regions of the Y-chromosome. The grey bars indicate the SNPs in all genomes and the black bars indicate the SNPs Number of occurrences of from Y-SNPsamples with an excellent quality, i.e. MCC ≥ 0.95. 2500 2000 A 1500 of Y-SNPs Number Whole Y-chromosome 1000 2500 500 2000 All SNPs 0 1500 Family-unique SNPs 1000 500 0 8-member family B Father-son pair Unique regions Y-chromosome 120 100 80 60 120 40 100 20 80 0 60 1 2 3 4 5 6 7 8 1 2 40 20 Number of occurrences of Y-SNP 0 Figure 6. Number of 1 new2Y-SNPs 3 per4 occurrence 5 6in the7 eight8paternally related 1 2 samples and the father-son pair of Complete Genomics: (A) SNPs in the whole Y-chromosome and (B) SNPs in only the ofidentified unique regions of the YNumber occurrences of Y-SNP chromosome. The grey bars indicate the SNPs found in the family. The black bars indicate the SNPs which are ‘family-unique’: they do not occur in any of the other samples with an excellent quality, i.e. MCC ≥ 0.95.