Methods Mice
advertisement

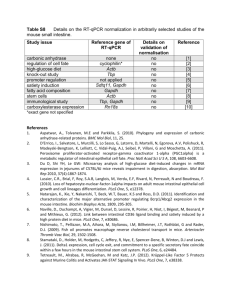
Methods Mice The recombination technique used in the Gba L444P mutation knock-in procedure, duplicated a part of the Gba gene. The resulting genomic structure was an intact Gba with the L444P mutation, a 3’ truncated copy which is a non-functional Gba gene missing upstream promotor sequences and 2 exons and, as reported by an intact metaxin gene (Liu at, 1998; Fig. 1B). Northern blots of mRNA from WT, Gba+/L444P and GbaL444P/L444P brains were probed with metaxin cDNA and the expression of the metaxin gene, was unaffected by the genomic changes in the Gba allele (Liu et al, 1998; Fig. 4A). In our hands, qPCR with metaxin assays showed no significant differences between GbaWT and Gba+/L444P (S4), consistent with Liu et al, 1998. Two independent PAC-Tg(SNCAA53T);Snca-/- lines expressing A53T mutant hSnca and lacking endogenous mSnca, were previously generated in the lab (Kuo et al. 2010). These were bred with heterozygous Gbatm1Rlp/+ to create a; PAC-Tg(SNCAA53T)+/-;Snca+/- ; Gbatm1Rlp/+mouse line. These lines were interbred to create a line of double transgenic animals, double-PAC-transgenic (SNCAA53T)+/+;Snca-/-; Gbatm1Rlp/+, homozygous for both insertion sites of the PAC and deficient in endogenous mSnca. The nomenclature for this line will be abbreviated throughout this paper to hSNCAA53T ;GbaL444P/+. Similarly, Gbatm1Rlp/+ was bred with PAC-Transgenic (SNCAWT)+/+;Snca-/- to obtain a Gbatm1Rlp/+; PAC-Tg(SNCAWT)+/+;Snca-/- stock. The nomenclature for this line will be abbreviated throughout this paper to hSNCAWT;GbaL444P/+. Homozygosity for the PAC was determined by interphase FISH using labeled SNCA on interphase peripheral blood mononuclear cells (Kuo et al. 2010) To reiterate, all transgenic lines, used in this study, are in an endogenous mSnca knock-out background . All non Gaucher mice, controls or wild types, were litter mates generated from the above crosses, and therefore share a similar genetic background (129S6/SvEvTac_FVB/N_C57/BL6), thus minimizing differences which could be due to the genetic background or to strain differences. A breeding scheme and a summary table describing the genotypes of the mice are found in (Supplementary Figure 1). Genotyping by PCR Genotyping of mouse tail genomic DNA was performed by published methods (Kuo et al. 2010). The L444P two bp mismatch at the mutation site was detected by primers using the mutant L444P allele 515 bp (cgtaactggatggacggact, Lmut-dF;acagtgctactgtttccgg, Lmut-dR). The primers used for the wild type specific allele of 1055 bp are gttccccaacacaatgctct, Lwt-dF and acagtgctactgtttccaa, Lwt-dR. Neuronal cultures: Cultures were prepared as previously described (Fishbein and Segal 2007), with modifications. P0-1 pups were genotyped, their brains removed to ice cold, oxygenated Leibovitz-15 medium (Invitrogen), supplemented with 0.5% glucose and Gentamicin (20ug/ml). The posterior one third of the cortices (including hippocampus) were separated, cut into pieces and incubated in 0.25% trypsin (UCSF Cell Culture Facility), 0.1% DNAse in Hank's Balanced Salt Solution (HBSS, Invitrogen), for 30 minutes at 37°C. Cortical tissue was washed in HBSS and mechanically dissociated by pipetting. About 2x106 living cells were plated per well of a 6 well plate. Plating medium consisted of Minimum Essential Media (MEM)-Earl salt (UCSF cell culture facility), supplemented with 0.6% Glucose, Gentamicin (20ug/ml-UCSF Cell Culture Facility), Glutamax, B-27 (Invitrogen) diluted 1/1000, 5% Fetal Bovine Serum and 5% Horse Serum (HS). Cells were left to grow in the incubator at 37°C, 5% CO2 for 4 days, medium was then changed to 10% HS in enriched MEM plus a mixture of 5'-fluoro-2deoxyuridine/uridine (20ug and 50ug /ml, respectively-Sigma). 4 days later, medium was replaced with 10% HS in enriched MEM, and half of it replaced again at the evening prior to metabolic labeling. Quantitative RT-PCR RNA was purified from neuronal cultures using the PureLink RNA mini kit (Ambion), according to manufacturer's directions. cDNA was prepared and RT-PCR preformed as previously described (Kuo, Li et al. 2010). Taqman Gene Expression assays( Life Technologies) were used: mSnca (Mm00447331_m1), synaptophysin (Mm00436850_m1) and Elavl4/HuD (Mm01263578_m1). The expression of mSnca was normalized to both synaptophysin and Elavl4. Also used were metaxin 1 (Mm01212769_g1), Gapdh (Mm99999915_g1 ) and actb (Mm00607939_s1).The expression of metaxin 1 was normalized to both gapdh and actin. The cycle number at which any particular sample crossed that threshold (Ct) was then used to determine fold difference. All the samples were measured in quadruplicate and averaged. Fold difference was calculated as 2ΔCt where Ct=[Ct(Syp)-Ct(SNCA)] or 2ΔCt where Ct=[Ct(Elavl4)Ct(SNCA)] and the expression relative to endogenous mouse Snca calculated as 2ΔΔCt where Ct =Ct(SNCA)- Ct(Snca). Western blotting Brain samples were prepared as described previously (Kuo et al. 2010). Neuronal cultures were homogenized in RIPA buffer (250μl/ 35mm plate). Cells membranes were mechanically disrupted by four cycles of 30 seconds vortex agitation and one minute incubation on ice, followed by 10min of sonication in ice cold water. Samples (from either brains or cultures) were centrifuged for 30 minutes to remove particulates. Total protein concentration was determined by the BCATM Protein Assay kit (Pierce). Lysates were mixed 1:1 (v/v) with 2X Laemmli loading buffer containing 10 % βME (Biorad), boiled and 15μg protein/well were subjected to SDS- PAGE (12% Mini-PROTEAN TGXTM Gels -BIO RAD). The following antibodies were used: mouse anti-Snca (BD Transduction Laboratories), mouse anti-phospho S129 Snca (Wako), rabbit anti-GCase (Sigma), mouse anti-tubulin (Calbiochem), anti-neuronal specific enolase (NSEAbcam), mouse anti-NeuN(Millipore) and goat anti-actin (Santa Cruz Biotech Inc). Horseradish peroxidase goat anti-mouse (GE Healthcare) or anti-rabbit (Cell Signaling) secondary antibodies and Clarity (BioRad) or Prime ECL kits were used. Analyses were done in Image-J software. Motor Tests Motor tests were carried out at two different ages (7 and 14 months old), using age-matched animals of both genders. Rotarod and open field test were done as previously described (Kuo et al. 2010) Colonic motility. Colonic motility was measured by the bead expulsion test as previously described. Briefly, a glass bead (diameter, 3 mm) was inserted through the anus and pushed, with a polished glass rod, into the colon for a distance of 2 cm. The time required for expulsion of the glass bead was recorded. Whole gut transit time Whole gut transit time was measured by oral gavage of 0.3 ml of 6% (w/v) carmine dye in 0.5% methylcellulose (Sigma) to each mouse as previously described. The time taken from the administration of carmine until the first appearance of one red fecal pellet was recorded. Immunocytochemistry and Immunohistochemistry Cultures: Neuronal-glial cultures were grown on 12 mm cover-slips, coated with Poly-L-Lysine (Sigma) and Laminin (Invitrogen). Cells were fixed in 4% w/v paraformaldehyde in PBS for 20 minutes, washed in PBS, blocked (1h) in 10% Goat Serum (GS) with 0.25% Triton-X, and overnight at 40C with primary antibodies (Mouse anti-MAP2, Sigma; Rabbit anti GFAP, Dako) in 2% GS, 0.1% Triton-X PBS. Coverslips were then washed in PBS and incubated for 30 min at room temp with secondary Alexa Fluor antibodies (Invitrogen), washed and mounted in Prolong Gold (Invitrogen). Brain sections were stained as previously described. (Kuo et al. 2010, Waxman et al. 2008, Waxman and Giasson 2008, Giasson et al. 2000). In short: Mice were anesthetized by intraperitoneal injection of Avertin, then intracardially perfused with PBS heparin (10u/ml) followed by 70% ethanol/ 150mM NaCl fixative. Brains were removed and post-fixed overnight at 4°C in fresh fixative. Immunohistochemistry was performed as described previously (Waxman et al. 2008a) specifically with pSer129/81A antibody. Western immunoblots showing the colocalization of phosphor-synuclein detected by the pSer129/81A antibody with synuclein, have been previously shown (Waxman & Giasson, 2010, Waxman & Giasson, 2008). In order to exclude the possibility of non-specific staining against phosphorelated neurofilament (NFL),which was previously observed to cross-react with the pSer129/81A antibody (Sacino, Brooks, et al., 2014), sections were stained with the following three different antibodies against NFL (Anti-NFL 68 /NR4 Sigma; NF-L (C28E10) #2837 Cell Signaling and NF-L, RPCA-NF-L EnCor Bio) . No NFL was detected by any of these antibodies in the structures positive with pSer129/81A staining. Analysis was done with Image scope software (Aperio) Immunoprecipitation The specificity of the α-synuclein band was confirmed in the following way: after immunoprecipitation with a rabbit antibody (Santa Cruz Biotech), proteins were transferred to a PDVF membrane for Western blot analysis and probed with a different antibody than the one used for immuno-precipitation (BD mouse monoclonal), which clearly recognized α-synuclein. In addition, the remaining gel was dried and autoradiography showed that the signal correlated with the Western blot signal. Gels were routinely silver stained prior to drying and phosphorimaging to ensure the clear presence of the α-synuclein. In the vast majority of cases, no non-specific signals were visible below 45 kDa, in neither silver stained gels nor phosphor imager readings. In the rare occasion that back ground noise was oberserved in the molecular weight range of αsynuclein, the data were discarded. Result S4 We preformed qPCR on our mice to look at metaxin expression in the new mouse stocks that were generated with the transgenic synuclein mice. We used a metaxin 1 expression assay (Mm01212769_g1) located at the 3’ end of the metaxin gene immediately adjacent to the 3’ end of Gba. We used cDNA transcribed from RNA from brains of hSncaA53T -GbaWT (n=3) and hSncaA53T -Gba+/L444P (n=3) litter mates and hSncaWT-GbaWT (n=3) and hSncaWT Gba+/L444P (n=2) litter mates. Controls used were Gapdh (Mm99999915_g1) and actinb (Mm00607939_s1). We found a 18-24% decrease in metaxin expression (compared to GbaWT ) that was not statistically significant (P for ct > 0.05) . We conclude that it is unlikely that a deficiency in the metaxin gene would be responsible for altering synuclein metabolism. Actin (reference gene) Gapdh (reference gene) hSncaA53T -Gba+/L444P -22% P(of ct) >0.05 compared to hSncaA53T -GbaWT -24% P(of ct) >0.05 hSncaWT Gba+/L444P compared to hSncaWT-GbaWT -23% P(of ct) >0.05 -18% P(of ct) >0.05