Manuscript: HBD-2 fusion proteins
advertisement

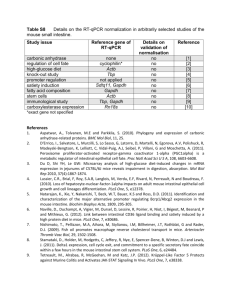
Supplemental Methods Quantification of HBD2-specific mRNA. Total RNA from HT-29 cells was prepared using RNApureTM according to manufacturer’s instructions (Peqlab) and mRNA from 1.25 µg total RNA was reversely transcribed using random hexamer and oligo(dT)15 (Promega). In the semiquantitative approach, standard PCR (Roche) was performed with 30 cycles from 62.5 ng cDNA using the specific primer pairs for HBD2. For HBD2, a second round of PCR using the same primer pair produced detectable amounts of the expected 195 bp fragment. PCR products were separated by agarose gel electrophoresis and visualized after ethidium bromide staining. Using the Image J software (National Institutes of Health), mean band densities representing the 195 bp (HBD2) and 422 bp PCR product (GAPDH) were evaluated. HBD2 expression was calculated as the quotient of the HBD2 and GAPDH band mean densities of each sample. RNA preparations were treated with RNase-free DNase (Sigma-Aldrich) before reverse transcription reaction in order to exclude relevant DNA contamination. PCR revealed comparable relations of HBD2 and GAPDH from untreated and DNAse treated templates (not shown). The TaqMan® gene expression assay for HBD2/DEFB4 (HS00823638m1) or the GAPDH (4326317E; both from Appleid Biosystems) comprising primer pairs matched for different genomic regions of two different exons, excluding bias of the genomic DNA amplification. Cloned HBD2 and the GAPDH fragment derived from the sequence of the respective gene expression assay cloned in pCR2.1-TOPO (Invitrogen) were used as standards (pHBD2 and pGAPDH). Quantification was optimized in the range of 102-109 template molecules per reaction using linearized plasmid DNA with 360 nM of the respective TaqMan® Gene expression assay for HBD2 and GAPDH in the StepOne Plus Thermocycler (Applied Biosystems; Supplemental Figure 1). HBD2 and GAPDH specific amplifications for individual samples from HT-29 cells were performed in PCR reactions containing both assay, and in parallel to mixed plasmid standards. Copy numbers of HBD2 or GAPDH mRNA were calculated from the CT values of the samples in relation to 1 the respective standard curve. HBD2 expression was calculated as the quotient of the copy numbers of HBD2 and GAPDH mRNA. Western-blot analysis. Detached HT-29 cells, preadipocytes or homogenized mucosal scrapings of human colon/ ileum sections were lysed with 3% SDS in 50 mM Tris-HC, pH 8.0 and immediately transferred to reducing sample buffer for SDS-PAGE. Heat denatured proteins were separated by gradient Tris-glycine SDS-PAGE (5-22.5% polyacrylamide). The following prestained markers were used to estimate protein sizes of the HBD2 peptide (about 4 kDa) and the larger fusion proteins (about 35 kDa): Color Marker Ultra-Low Range 1-26 kDa (Sigma-Aldrich) and Page RulerTM Prestained Protein Ladder 11-170 kDa (MBI Fermentas). Proteins were transferred to a polyvinylidene fluoride membrane with a pore size of 0.22 µm (Millipore) by tank blotting (Biometra). Air-dried membranes were incubated overnight with 1% protein-free reagent (Roche) in TBST buffer containing 50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.1% Tween-20 (Sigma-Aldrich). After extensive washing with TBST, membranes were incubated for 2 h with the polyclonal goat-derived HBD2-specific antibody (200 ng/mL; sc10854; Santa Cruz Biotechnology) diluted in TBST. Membranes, again washed twice with TBST, were incubated for 1.5 h with a horseradish peroxidase-coupled secondary goat-specific antibody (16 ng/mL; 705035147, Jackson ImmunoResearch). The primary antibody for protein loading control was the murine monoclonal antibody specific to human β-actin (1:10.000; clone AC-15, Sigma-Aldrich) followed by a horseradish peroxidase-coupled secondary mouse-specific antibody (1:1000, P0161, Dako). Antibody binding was detected using the ECLTM system (Roche). Chemiluminescence was visualized by LAS 1000 (Fujifilm Life Sciences). Exposition times were 10-30 min for HBD2 and 10 sec-1 min for β-actin. Cloning of HBD2 for eukaryotic and prokaryotic fusion protein expression. 2 HT-29 cells were treated with 10 ng/mL IL-1β for 6 h and cDNA was prepared. Appropriate terminal restriction sites, inserted by PCR primers, allowed directed cloning of HBD2 sequences. A complete HBD2 mRNA for constitutive eukaryotic expression of a secreted HBD2-EGFP fusion protein was amplified using the primer pair 5’-GCTCCCAGCCTCGAGCCATGAGGGTC-3’/ 5’-GCTTCTTGGCCGAATTCTGGCTTTTTGC-3’. Using standard recombinant techniques including sequence verification (Seqlab), the HBD2 construct was cloned as an XhoI/EcoRI fragment into the vector pEGFP-N2 (pEGFP; Clontech) for expression under the control of the early promoter/enhancer of the human cytomegalovirus (pCMV). The resulting pHBD2 vector contained the native HBD2 leader-sequence for secretion of the HBD2-EGFP fusion protein by transfected mammalian cells. For prokaryotic expression, a cDNA for mature HBD2 was recovered with the primer pair 5’-CTCTTCCAGGTGGATCCGGTGGTATAGG-3’/ 5’-GCTTCTTGGCCGAATTCTGGCTTTTTGC-3’ and cloned as a BamHI/EcoRI fragment downstream of the GST sequence into the vector pGEX-2T (GE Healthcare). For the controlled fusion protein production and subsequent HBD2 release, the isopropylbeta-D-thiogalactopyranoside (IPTG) inducible lactose operon promoter (plac) and the internal thrombin cleavage site (TCS) were used. Production and purification of GST-HBD2. E. coli BL21 were transformed with the expression vector for GST or for the GST-HBD2 fusion protein and cultured in LB medium. After initial culture to OD600nm of 0.4-0.5 (GST) or 1.0-1.2 (HBD2-GST) recombinant protein production was induced by 200 mg/mL IPTG for 4-5 h (GST) or 2 h (HBD2-GST). All following steps were performed on ice and with pre-cooled solutions and devices. Bacteria were collected by centrifugation (4,000xg, 20 min, 4°C, without brake) and resuspended in phosphate-buffered saline, pH 7.2 (PBS). Cells were lysed by low energy ultrasonic treatment (Bandelin Electronic) in the presence of 2 µg/mL DNase I and 50 µg/mL lysozyme (both from Fluka). Clear bacterial lysates 3 after high-speed centrifugation (12,000xg, 30 min, 4°C) were subjected to GST TrapTM affinity chromatography (GE Healthcare) and eluted with a buffer containing 50 mM Tris-HCl, pH 8.0, 100 mM NaCl and 20 mM gluthathione. If applicable, glutathione was removed by standard Sephadex G-25 gel filtration (GE Healthcare) and buffer was replaced for PBS or 50 mM Tris-HCl, pH 8.0. Protein concentrations of glutathione-free GST or GST-HBD2 preparations were determined using the bicinchoninic-acid assay (Pierce). Product purity was controlled by SDS-PAGE and staining with Coomassie brilliant blue-R250 (Merck) and HBD2 specific western-blot analysis. Final preparations to be used for the experiments were adjusted to 2.5 mg/mL, shock-frozen over liquid nitrogen and stored at -80°C. 4