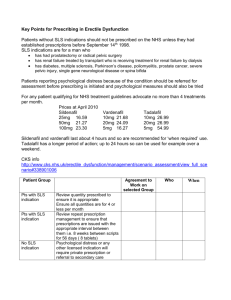
RAR - جامعة الكوفة - كلية الصيدلة
advertisement

A STUDY OF THE EFFECTS OF SILDENAFIL AND VARDENAFIL ON AORTIC INTIMA - MEDIA THICKNESS AND BLOOD FLOW VELOCITIES OF AORTA, RENAL ARTERY AND INTRA RENAL ARTERY IN HYPERLIPIDEMIC MALE RABBITS A THESIS Submitted To The College Of Medicine And The Committee Of Postgraduate Studies Of Kufa University In Partial Fulfillment Of The Requirements For The Degree Of Master Of Science In Pharmacology And Therapeutics BY Karrar Hussein Kammona B.Sc. Pharmacy Supervised by Dr. Najah R. AL-Mousawi Professor of Clinical Pharmacology and Therapeutics Ph.D,FRCP,FACP 1430 A.H Dr. Akeel AM. H. Zwain Lecturer of Cardiovascular Physiology Ph.D U.K. 2009 A.D. We certify that this thesis was prepared under our supervision at the College of Medicine, University of Kufa , as a partial requirement for the Degree of Master of science in Pharmacology and Therapeutics. Signature ………….. Signature…………… Name: Najah R. AL-Mousawi Name: Akeel AM. H. Zwain Professor of Clinical Pharmacology and Therapeutics Ph.D, FRCP, FACP Supervisor Lecturer of Cardiovascular Physiology Ph.D U.K. Supervisor In view of the available recommendations . I put forward this thesis for debate by the examining committee . Signature…………… Name: Najah R. AL-Mousawi Professor of Clinical Pharmacology and Therapeutics Head Department of Pharmacology and Therapeutics Acknowledgement Praise be to our Almighty Allah, the Gracious who gives me the power and motivation to perform and present this work. I wish to express my heartfelt gratitude and appreciation to my honorable supervisor Prof. Dr. Najah R. Al Mousawi for his guidance, valuable advices and support. I wish to address special thanks to Dr. Akeel AM.H. Zwain, my honorable supervisor for his great help and support in the field of Doppler ultrasound examinations and measurements . I am deeply grateful to Kufa College of Medicine, represented by the Dean and the staff for providing facilities required for this work. I deeply appreciate Dr. Hussein Abed Al-khahdam for his kind help. I am so thankful to Assistant prof. Dr. Majed K. Hussein & Dr Ahmed Al-Mohana for their kind help. Also I would like to express my deepest thanks to Dr. Liwaa H. Mahdi AlKulabi who help me in the field of histopathology. I highly appreciate Assistant prof. Dr Abdul-Kareem Al-Mayah for his help in statistical analysis. My gratitude and appreciation to pharmacist Muhammad Ali & Hayder Chassep for their kind helps. I am grateful to Mr. Fares Abbas for his help in blood sampling. Finally, my heartfelt appreciation and gratitude to My Family for their infinite support and patience during the time of the study. Karar H. Kammona List of Contents Subject Page Dedication І Acknowledgments ІІ List of Contents ІІІ List of tables IX List of figures XІІ List of abbreviations XVІ Summary XIX Chapter One: Introduction and Literature Review 1.1 Atherosclerosis 1 1.1.1 Mortility and morbidity 1 1.1.2 Risk factor of atherosclerosis 2 1.1.3 Pathogenesis of atherosclerosis 2 1.1.3.1 Hypotheses of atherogenesis 2 1.2 Oxidative Stress 6 1.2.1 Free radicals 6 1.2.2 Oxidative stress and reactive oxygen species 7 1.2.3 Sources and reactions of ROS and RNS 7 1.2.4 Antioxidants 8 1.2.5 Oxidative stress in atherosclerosis 9 1.3 Endothelium dysfunction 10 1.3.1 Endothelial dysfunction as an initial step of atherosclerosis 10 1.3.2 Oxidative stress and endothelial dysfunction 11 1.3.3 Antioxidant properties of nitric oxide in atherosclerosis 12 1.4 Treatment of Atherosclerosis 13 1.4.1 Lifestyle change 13 1.4.2 Medications 13 1.4.3 Therapeutic approach 14 1.5 Phosphodiesterase Inhibitors. 15 1.5.1 Sildenafil (Viagra) 15 1.5.1.1 Mechanism of action and pharmacological properties 15 1.5.1.2 Effect on hemodynamics 16 1.5.1.3 Pharmacokinetics and metabolism 17 1.5.1.4 Adverse effects 18 1.5.1.5 Drug interaction 18 1.5.2 Vardenafil (Levitra®) 18 1.5.2.1 Mechanism of action and pharmacological properties 19 1.5.2.2 Hemodynamic effects 19 1.5.2.3 Pharmacokinetics 20 1.5.2.4 Side effects 21 1.5.2.5 Drug interaction 21 1.6 Doppler ultrasound technique 21 1.6.1 Doppler parameters and Doppler indices 22 1.7 Blood flow rate and blood flow velocity 24 1.7.1 Definition of blood flow and blood flow velocity 24 1.7.2 Factors affecting on blood flow and blood flow velocity 24 1.7.3 Effect of atherosclerosis on blood flow velocity 25 1.7.4 Aortic blood flow 26 1.7.5 Renal artery blood flow 26 1.7.6 Intra-renal arteries blood flow 27 1.8 Aim of the study 28 Chapter Two: Material and Methods 2.1 Materials 29 2.2 Animals and study design 30 2.3 Drugs 32 2.3.1 Sildenafil (viagra) 32 2.3.2 Vardenafil (levatra) 32 2.4 Preparation of sample 32 2.4.1 Blood sampling 32 2.4.2 Tissue sampling 32 2.5 Measurement of samples 34 2.5.1 Measurement of serum total cholesterol 34 2.5.2 Measurement of Triglycerides Concentration 35 2.5.3 Measurement of HDL-C Concentration 37 2.5.4 Calculation of LDL,VLDL and Atherogenic index 38 2.5.5 Measurement of serum malondialdehyde (MDA) Level 38 2.5.6 Determination of serum reduced glutathione (GSH) 39 2.6 Measurement of aortic diameter, intima-media thickness and blood flow velocities of the aorta, renal artery and intra-renal arteries 42 2.7 Calculation of Doppler indices 43 2.8 Histopathological procedure 43 2.9 Statistical analysis 44 Chapter Three: Results 3.1 Effect on serum total cholesterol (TC) level 45 3.2 Effect on serum triglyceride (TG) level 46 3.3 Effect on HDL-C level 48 3.4 Effect on serum LDL-C level 49 3.5 Effect on VLDL-C level 51 3.6 Effect on atherogenic index 52 3.7 Effect on serum level of Malon Dialdehyde MDA . 54 3.8 Effect on serum reduced glutathione level GSH . 55 3.9 Effect on aortic intima-media thickness (IMT) 57 3.10 Effect on aortic diameter 58 3.11 Effect on aortic peak systolic blood flow velocity (PSV) 60 3.12 Effect on aortic end diastolic blood flow velocity (EDV) 61 3.13 Effect on aortic blood flow resistive index (RI) 63 3.14 Effect on aortic blood flow pulsatality index (PI) 64 3.15 Effect on renal artery peak systolic blood flow velocity(PSV) 66 3.16 Effect on renal artery end diastolic blood flow velocity(EDV) 67 3.17 Effect on renal artery blood flow resistive index (RI) 69 3.18 Effect on renal artery blood flow pulsatality index (PI) 70 3.19 Effect on intra-renal arteries peak systolic blood flow velocity 72 3.20 Effect on intra-renal arteries end diastolic blood flow velocity 73 3.21 Effect on intra-renal arteries blood flow resistive index (RI) 75 3.22 Effect on intra-renal arteries blood flow pulsatality index (PI) 76 3.23 Effect on renal artery-to-aortic peak systolic velocity ratio 78 3.24 Histopathological findings 79 Chapter Four: Discussion 4.1 Effect on Serum Lipid Profile 89 4.1.1 Effect of cholesterol enriched diet on serum lipid profile 89 4.1.2 Effect of Sildenafil and Vardenafil treatment on serum lipid profile 89 4.2 Effect on oxidative stress 89 4.2.1 Effect of high cholesterol diet on oxidative stress 89 4.2.2 Effect of Sildenafil and Vardenafil on MDA and GSH level 90 4.3 Effect on aortic intima-media thickness 91 4.3.1 Effect of cholesterol enriched diet on aortic intima-media thickness 91 4.3.2 Effect of Sildenafil and Vardenafil treatment on aortic intimamedia thickness 91 4.4 Effect on aortic diameter 92 4.4.1 Effect of cholesterol enriched diet on aortic diameter 92 4.4.2 Effect of Sildenafil and Vardenafil treatment on aortic diameter 93 4.5 Effect on peak systolic blood flow velocity 93 4.5.1 Effect of cholesterol enriched diet on peak systolic velocity 93 4.5.2 Effect of Sildenafil and Vardenafil treatment on peak systolic velocity 94 4.6 Effect on End diastolic blood flow velocity 95 4.6.1 Effect of cholesterol enriched diet on end diastolic velocity 95 4.6.2 Effect of Sildenafil and Vardenafil treatment on end diastolic velocity 96 4.7 Effect on resistive index (RI) 96 4.7.1 Effect of cholesterol enriched diet on resistive index 96 4.7.2 Effect of Sildenafil and Vardenafil treatment on resistive index 97 4.8 Effect on pulsatality index (PI) 98 4.8.1 Effect of cholesterol enriched diet on pulsatality index 98 4.8.2 Effect of Sildenafil and Vardenafil treatment on pulsatality index 98 4.9 Effect on renal artery-to-aortic peak systolic velocity ratio 99 4.9.1 Effect of cholesterol enriched diet on renal artery –to-aortic peak systolic velocity ratio (RAR) 99 4.9.2 Effect of Sildenafil and Vardenafil treatment on renal artery – to-aortic peak systolic velocity ratio (RAR) 99 4.10 Effect on aortic histological change 100 4.10.1 Effect of cholesterol enriched diet on aortic histology 100 4.10.2 Effect of Sildenafil and Vardenafil on aortic histology 100 Chapter five : Conclusions and Recommendations 5.1 Conclusions 101 5.2 Conclusions Recommendations 102 References References 103 List of Tables Table Page Table (1): list of chemicals, pharmaceuticals, reagent and their supplier 29 Table (2): Instrument and their supplier 30 Table (3): Changes of serum TC level in mg/dl of the four experimental groups 45 Table(4): Multiple comparisons between different groups mean values of serum TC level in (mg/dl). Table(5): Changes of serum TG level in mg/dl of the four experimental groups. 45 Table(6): Multiple comparisons between different groups mean values of serum TG level in ( mg/dl) 47 Table (7): Changes of serum HDL-C level in mg/dl of the four 48 47 experimental groups. Table (8) : Multiple comparisons between different groups mean values of serum HDL-C level in ( mg/dl) . Table (9): Changes of serum LDL-C level in mg/dl of the four experimental groups. Table (10) : Multiple comparisons between different groups mean values of serum LDL-C level in ( mg/dl) . Table (11): Changes of serum VLDL-C level in mg/dl of the four experimental groups. Table (12) : Multiple comparisons between different groups mean values of VLDL-C level . Table(13): Changes of atherogenic index experimental groups. in of the four 48 50 50 51 51 53 Table (14) : Multiple comparisons between different groups mean values of atherogenic index . 53 Table (15): Sequential changes of serum MDA level mmole/L of the four experimental group. 54 Table (16): Multiple comparisons among different groups mean values of serum MDA level mg/dl . 54 Table (17): Sequential changes of serum GSH level uM of the four experimental groups. 56 Table (18): Multiple comparisons among different groups mean values of serum GSH level . 56 Table (19): Effect of cholesterol enriched diet , Vardenafil and Sildenafil treatment on the rabbits aortic intima-media thickness . 57 Table (20) : Multiple comparisons between different groups mean values of rabbits aortic intima-media thickness . 57 Table (21): Effect of cholesterol enriched diet , vardenafil and sildenafil treatment on rabbits aortic diameter . 59 Table (22): Multiple comparisons between different groups mean values of rabbits aortic diameter . 59 Table (23): Effect of cholesterol enriched diet , vardenafil and sildenafil treatment on the rabbits aortic peak systolic blood flow velocity . Table (24): Multiple comparisons between different groups mean values of rabbits aortic peak systolic blood flow velocity. Table (25): Effect of cholesterol enriched diet , vardenafil and sildenafil treatment on the rabbits aortic end diastolic blood flow velocity . Table (26): Multiple comparisons between different groups mean values of rabbits aortic end diastolic blood flow velocity . 60 60 62 62 Table (27): Effect of cholesterol enriched diet , vardenafil and sildenafil treatment on the rabbits aortic blood flow resistive index . 63 Table (28): Multiple comparisons between different groups mean values of rabbits aortic blood flow resistive index . 63 Table (29): Effect of cholesterol enriched diet , , vardenafil and sildenafil treatment on the rabbits aortic blood flow pulsatality index . 65 Table (30): Multiple comparisons between different groups mean values of rabbits aortic blood flow pulsatality index . 65 Table (31): Effect of cholesterol enriched diet , vardenafil and sildenafil treatment on the rabbits renal artery peak systolic velocity. 66 Table (32): Multiple comparisons between different groups mean values of rabbits renal artery peak systolic blood flow velocity . 66 Table (33): Effect of cholesterol enriched diet , , vardenafil and sildenafil treatment on the rabbits renal artery end diastolic velocity . 68 Table (34): Multiple comparisons between different groups mean values of rabbits renal artery end diastolic blood flow velocity . Table (35): Effect of cholesterol enriched diet , , vardenafil and sildenafil treatment on the rabbits renal artery blood flow resistive index . 68 69 Table (36): Multiple comparisons between different groups mean values of rabbits renal artery blood flow resistive index . 69 Table (37): Effect of cholesterol enriched diet , , vardenafil and sildenafil treatment on the rabbits renal artery blood flow pulsatality index . 71 Table (38): Multiple comparisons between different groups mean values of rabbits renal artery blood flow pulsatality index . 71 Table (39): Effect of cholesterol enriched diet , , vardenafil and sildenafil treatment on the rabbits intra- renal arteries peak systolic velocity . 72 Table (40): Multiple comparisons between different groups mean values of the rabbits intra- renal arteries peak systolic velocity . 72 Table (41): Effect of cholesterol enriched diet , vardenafil and sildenafil treatment on the rabbits intra- renal arteries end diastolic velocity . 74 Table (42): Multiple comparisons between different groups mean values of rabbits intra- renal arteries end diastolic velocity . 74 Table (43): Effect of cholesterol enriched diet , vardenafil and sildenafil treatment on the rabbits intra-renal arteries blood flow resistive index . 75 Table (44): Multiple comparisons between different groups mean values of rabbits intra-renal arteries blood flow resistive index . 75 Table (45): Effect of cholesterol enriched diet , , vardenafil and sildenafil treatment on the rabbits intra-renal arteries blood flow pulsatality index . 77 Table (46): Multiple comparisons between different groups mean values of rabbits intra-renal arteries blood flow pulsatality index . 77 Table (47): Effect of cholesterol enriched diet , , vardenafil and sildenafil treatment on rabbits renal artery-to-aortic peak systolic velocity ratio . 78 Table (48): Multiple comparisons between different groups mean values of rabbits renal artery-to-aortic peak systolic velocity ratio . 78 Table (49): Demonstration of different aortic atherosclerotic phases for different rabbit group at the end of 12 weeks of the study 80 Table (50): Demonstration of different aortic atherosclerotic lesions for different rabbit group at the end of 12 weeks of the study 81 List of Figures Figure Page Figure (1): Effect of Vardenafil and Sildenafil on serum TC level in comparison to the two control groups (normal and induced untreated). 46 Figure(2): Effect of vardenafil and sildenafil treatment on serum TG level in comparison to the two control groups ( normal and induced untreated. 47 Figure (3): Effect of sildenafil 5mg/kg/day and verdenafil 18mg./kg/day treatment on serum HDL-C level in comparison to the two control group (normal and induced untreated). 49 Figure (4): Effect of vardenafil and sildenafil treatment on serum LDL-C level in comparison to the two control groups (normal and induced untreated) 50 Figure (5): Effect of vardenafil and sildenafil treatment on serum VLDL-C level in comparison to the two control groups (normal and induced untreated) 52 Figure (6): Effect of vardenafil and sildenafil treatment on atherogenic index in comparison to the two control group ( normal and induced untreated) 53 Figure (7) : The change in MDA Level in the four experimental group 55 Figure (8): The change in Serum GSH Level in the four experimental groups 56 Figure (9): Effect of Vardenafil and Sildenafil treatment on rabbits aortic intima-media thickness (IMT) measured in (mm) in comparisons to the two control group (normal and 58 induced untreated) Figure (10): Effect of vardenafil and sildenafil treatment on rabbits aortic diameter measured in (mm) in comparisons to the two control group (normal and induced untreated) 59 Figure (11): Effect of vardenafil and sildenafil treatment on rabbits aortic peak systolic velocity (PSV) measured in (cm/s) in comparisons to the two control group ( normal and induced untreated) 61 Figure (12): Effect of vardenafil and sildenafil treatment on rabbits aortic end diastolic velocity (EDV) measured in (cm/s) in comparisons to the two control group ( normal and induced untreated) 62 Figure (13): Effect of vardenafil and sildenafil treatment on rabbits aortic blood flow resistive index (RI) in comparisons to the two control group (normal and induced untreated) 64 Figure (14): Effect of vardenafil and sildenafil treatment on rabbits aortic blood flow pulsatality index (PI) in comparisons to the two control group (normal and induced untreated) 65 Figure (15): Effect of vardenafil and sildenafil treatment on rabbits renal artery peak systolic velocity (PSV) measured in cm/s in comparisons to the two control group (normal and induced untreated) 67 Figure (16): Effect of vardenafil and sildenafil treatment on rabbits renal artery end diastolic velocity (EDV) measured in cm/s in comparisons to the two control group (normal and induced untreated) 68 Figure (17): Effect of vardenafil and sildenafil treatment on rabbits renal artery blood flow resistive index (RI) in comparisons to the two control group (normal and 70 induced untreated) Figure (18) : Effect of vardenafil and sildenafil treatment on rabbits renal artery blood flow pulsatality index (PI) in comparisons to the two control group (normal and induced untreated) 71 Figure (19): Effect of vardenafil and sildenafil treatment on the rabbits intra- renal arteries peak systolic velocity (PSV) measured in cm/s in comparisons to the two control group (normal and induced untreated) 73 Figure (20): Effect of vardenafil and sildenafil treatment on rabbits intra- renal arteries end diastolic velocity (EDV) measured in cm/s in comparisons to the two control group (normal and induced untreated) 74 Figure (21): Effect of vardenafil and sildenafil treatment on rabbits intra-renal arteries blood flow resistive index (RI) in comparisons to the two control group (normal and induced untreated) 76 Figure (22): Effect of vardenafil and sildenafil treatment on rabbits intra-renal arteries blood flow pulsatality index (PI) in comparisons to the two control group (normal and induced untreated) 77 Figure (23): Effect of vardenafil and sildenafil treatment on rabbits renal artery-to-aortic peak systolic velocity ratio in comparisons to the two control group (normal and induced untreated) 79 Figure (24): Percentages of aortic involvement with different atherosclerotic lesion phases 81 Figure (25): Percentages of aortic involvement with different atherosclerotic lesions. 82 Figure (26): A cross section of normal rabbit aorta shows the normal appearance of arterial wall 82 Figure (27): A cross section of hypercholesterolemic rabbit's aorta demonstrating appearance of lipid laden macrophages (Foam cells) . 83 Figure (28): A cross section of hypercholesterolemic rabbit's aorta demonstrating many lipid laden macrophages (Foam cells) . 83 Figure (29): A cross section from aorta shows a pathological intimal thickening with a poorly formed fibrous cap. 84 Figure (30): A cross section from aorta shows a pathological intimal thickening with a poorly formed fibrous cap. The section stained with haematoxylin and eosin ( ×10) 84 Figure (31): A cross section of hypercholesterolemic rabbit's aorta demonstrating endothelial cell with underlying Foam cells . Intial lesion (phase 2 atherosclerosis). The section stained with haematoxylin and eosin ( ×40) 85 Figure (32): A cross section from aorta shows a fibro-atheromatous plaque with a thin layer of fibrous connective tissue overlying a largely necrotic, fatty mass. Advance atherosclerotic lesion (phase V). The section stained with haematoxylin and eosin ( ×10) 85 Figure (33): A B-mode ultrasound image showing a rabbit abdominal aorta with normal aortic wall thickness , D1 is the intima-media thickness = 0.3 mm , D2 is the aortic diameter = 2.1 mm 86 Figure (34): A B-mode ultrasound image showing a rabbit abdominal aorta with an increased aortic thickness (sclerotic wall), but there is no apparent plaques , D1 is the intima-media thickness = 0.6 mm , D2 is the aortic diameter = 2.8 mm 86 Figure (35):AB-mode ultrasound image showing a rabbit abdominal aorta with an increased aortic thickness (sclerotic wall), there also dissemination of plaques inside the aorta , D1 is the intima media thickness = 0.7 mm 87 Figure(36): A Doppler wave form spectrum showing a rabbits aortic blood flow velocity, PSV=0.49 m/s , EDV=0.06 m/s RI=0.88 , S/D ratio=8.2 87 Figure (37): A Doppler wave form spectrum showing a rabbits renal artery blood flow velocity, PSV=0.48 m/s , EDV=0.17m/s, RI=0.58 , S/D ratio=2.4 88 Figure(38): Doppler wave form spectrum showing a rabbits intrarenal artery blood flow velocity, PSV=0.49 m/s , EDV=0.20m/s, RI=0.59 , S/D ratio=2.5 88 List of Abbreviations Abbreviations Meaning ACE inhibitor Angiotonsine converting enzyme inhibnitor Ach Acetylcholine ANOVA Analysis of ovariance BP Blood pressure BT Bleeding time CAD Coronary Artery Disease cAMP Cyclic adenosine mono phosphate cGMP Cyclic Guanosine Monophosphate CRP C-Reactive protein DTNB 5,5-dithiobis (2-nitrobenzoic acid) e NOs Endothelial nitric oxide synthase ED Erectile dysfunction EDCFs Endothelium-derived contracting factors EDRFs Endothelium-derived relaxing factors EDTA Ethylenediamine tetra-acetic acid EDV End diastolic velocity ET-1 Endotheline-1 FDA Food and Drug Administration GPx Glutathione peroxidase GSH Glutathione GssG Oxidized Glutathione GTP Guanine triphosphate HDL High density lipoprotein HF Heart failure HMG-CO-A 3-Hydroxy-3-methylglutaryl coenzyme A HR Heart rate IC50 Half-maximal inhibition ICAM Inter cellular adhesion molecules LDL Low density lipoprotein LPL Lipoprotein lipase MDA Malondialdehyde MMPs Matrix metalloproteinases MV Mean velocity NADPH Nicotinamide Adenine Dinucleotide Phosphate NAION Non-arteritic ischaemic optic neuropathy NO Nitric Oxide oxLDL Oxidized LDL PAP Pulmonary artery pressure PDEIs Phosphodiesterase enzyme inhibitors PDEs Phosphodiesterase enzymes PH Pulmonary hypertension PI Pulsatality index PMNs Polymorphonuclear leukocytes PPAR Peroxisome proliferator activated receptor PSV Peak systolic velocity RAR Renal artery-to Aortic peak systolic velocity Ratio RAS Renal artery stenosis RI Resistive index RNS Reactive nitrogen species ROS Reactive Oxygen species RVSP Right ventricular systolic pressure S/D ratio Systolic velocity/ Diastolic velocity ratio SEM Standard error of mean SMCs Smooth muscle cells SOD Superoxide dismutase TAB Thiobarbituric TC Total cholesterol TCA Trichloroacetic acid TG Triglyceride VCAM Vascular cellular adhesion molecules VLDL Very low density lipoprotein XO Xanthine oxidase Summary Atherosclerosis is the major world wide killing disease. The most common risk factors are hyperlipidemia, diabetes, arterial hypertension, cigarette smoking and obesity. PDEIs (sildenafil and vardenafil) are the main drugs used in the treatment of erectile dysfunction, and promise in treatment of atherosclerosis. Method : Twenty four local domestic male rabbits were involved in this study. The animals were randomly divided in to four groups , Group I rabbits fed normal chow (oxiod) diet for 12 weeks. Group II rabbits fed 2% cholesterol enriched diet for 12 weeks. Group III rabbits fed with cholesterol enriched diet for 6 weeks, and then continued on cholesterol enriched diet and treated with sildenafil 5mg./kg./day orally for the next 6 weeks. Group IV rabbits fed with cholesterol enriched diet for 6 weeks ,and then continued on cholesterol enriched diet and treated with vardenafil 18mg./kg./day orally for the next 6 weeks.The blood samples were collected at the start of the study, 6weeks of the study and then every week through the 6 weeks of treatment course. Serum lipids profile [Total cholesterol (TC), Triglyceride (TG), High density lipoprotein (HDL), Low density lipoprotein (LDL),Very low density lipoprotein (VLDL)] ,atherogenic index and oxidative stress parameters [GSH,MDA] were measured. Aortic diameter and intima- media thickness and aortic, renal artery and intra-renal artery blood flow velocities [ Peak systolic velocity (PSV), End diastolic velocity(EDV), Pulsatality index (PI), Resistive index (RI), and Renal artery -to- Aortic peak systolic velocity Ratio (RAR)] were measured by triplex Doppler machine at the start of the study, 6weeks, and then at the end of the study. Autopsy of aortic sectioning for histopathology was done at the end of the study. Results: 2%cholesterol enriched diet results in significant increase (P<0.05) in serum level of TC, TG, HDL, LDL, VLDL, atherogenic index with decrease serum GSH level and significant increase (P<0.05) in serum MDA level. Also there was asignificant increase (P<0.05) in aortic diameter , intima-media thickness, PI, RI, renal artery PSV, PI, RI, intra-renal artery PSV, PI, and RI in comparison to the normal control group. Also there was significant increase (P<0.05) in Renal artery -to- Aortic peak systolic velocity Ratio (RAR). There was no significant change (P>0.05) in aortic PSV and EDV, and no significant change (P>0.05) in renal artery and intra-renal artery EDV. In regard to histopathological results, all rabbits fed cholesterol enriched diet show development of different phases of atherosclerosis and there was significant difference (P<0.05) between the induced untreated group and the normal control group. Treatment of rabbits with sildenafil and vardenafil results in non significant reduction (P<0.05) in serum level of TC, TG,HDL, LDL, VLDL, and atherogenic index, significant reduction (P<0.05) in MDA level and significant increase (P<0.05) in serum GSH level. There was non significant reduction (P<0.05) in aortic diameter and intima-media thickness. Also there was significant reduction (P<0.05) in renal artery and intra-renal artery PSV, EDV, PI and RI. There was no significant change ( p>0.05) in aortic PSV, EDV, PI , RI and in Renal artery -to- Aortic peak systolic velocity Ratio (RAR) in comparison to the rabbits in the induced untreated control group. Regarding histopathological results, sildenafil and vardenafil treated rabbits showed atherolytic effect, but this effect did not reach the significant level (P>0.05) in comparison to the induced untreated group. There was no significant difference (P>0.05) between sildenafil treated group and vardenafil treated group in measured parameters. Conclusions: Sildenafil and vardenafil do not affect serum level of TC, TG, LDL, VLDL, HDL and atherogenic index, but they increase serum MDA level and decrease serum GSH level. Also both drugs insignificantly reduce severity of atherosclerosis and reduce intima- media thickness of arteries in treated animals. Therefore, both drugs diminish the arterial stiffness as indicated by the reduction of PI and RI of abdominal aorta, and reduction of PSV, EDV, PI and RI of renal artery and intra-renal arteries of the treated animals. 1.1 Atherosclerosis Atherosclerosis is a disease of large and medium-sized muscular arteries and is characterized by endothelial dysfunction, vascular inflammation, and the buildup of lipids, cholesterol, calcium, and cellular debris within the intima of the vessel wall. This buildup results in plaque formation, vascular remodeling, acute and chronic luminal obstruction, abnormalities of blood flow, and diminished oxygen supply to target organs(1). 1.1.1 Mortility and morbidity Arteriosclerosis of the coronary and peripheral vasculature is the leading cause of death among men and women in the United States and worldwide(2).Estimates for 2005 in the United States alone are that about 16 million people have atherosclerotic heart disease and 5.8 million have stroke. Cardiovascular disease, primarily coronary and cerebrovascular atherosclerosis, caused almost 870,000 deaths in 2005—almost twice as many as cancer caused and 9 times as many as injuries caused(3). The incidence of all forms of atherosclerosis increases with age even among the elderly. However, during the last 30 years, the mortality rate for atherosclerosis has markedly and progressively decreased in the USA and in several other industrialized countries. The decreasing mortality rate is probably primarily due to the widespread use of preventive strategies, resulting in decreased prevalence of such risk factors as untreated hypertension, abnormal lipid levels, and cigarette smoking. Improvements in diagnosis and treatment of established atherosclerosis probably have contributed less(4). 1.1.2 Risk factor of atherosclerosis A number of risk factors are associated with cardiovascular disease and these may be classified into two categories: fixed and modifiable. Fixed risk factors include genetic composition, age, menopausal status, and gender. The modifiable factors are a series of environmental cues and lifestyle choices including (but not limited to) diet, smoking, status of concurrent diseases (e.g. diabetes), exercise and ethanol consumption. When considering these risk factors it is noteworthy that many, if not all, contribute to disease progression at least in part via oxidative stress (5). 1.1.3 Pathogenesis of atherosclerosis 1.1.3.1 Hypotheses of atherogenesis Over the past 150 years, there have been numerous efforts to explain the complex events associated with the development of atherosclerosis. In this endeavor, three distinct hypotheses have emerged that are currently under active investigation. These hypotheses of atherosclerosis are not mutually exclusive but rather emphasize different concepts as the necessary and sufficient events to support the development of atherosclerotic lesions. For the purposes of this review, we refer to these paradigms as follows: 1) the response-to-injury, 2) the response-to-retention, and 3) oxidative modification (6). In response-to-injury hypothesis, the proposed initial step in atherogenesis is endothelial denudation leading to a number of compensatory responses that alter the normal vascular homeostatic properties. For example, injury enhances endothelial adhesiveness for leukocytes and platelets and alters the local vascular anticoagulant milieu to a procoagulant one. Recruited leukocytes and platelets then release cytokines, vasoactive agents, and growth factors that promote an inflammatory response that is characterized by migration of smooth muscle cells into the intima and their proliferation to form an intermediate lesion(7). Another inflammatory response is the recruitment of macrophages into the arterial wall which take up deposited LDL lipid to form lipid-laden "foam cells," the hallmark of an early atherosclerotic lesion. The process of lipid accumulation and foam cell formation perpetuates an inflammatory response that perpetuates macrophage and lymphocyte recruitment (8,9). Continued inflammation allows for cellular necrosis with a concomitant release of cytokines, growth factors, and proteolytic enzymes that set the stage for autocatalytic expansion of the lesion to form a space-occupying collection in the intima not unlike an abscess that would form in other tissues. As the lesion enlarges, it begins to encroach upon the lumen and, ultimately, blood flow is impaired(10). This response-to-injury hypothesis was originally based on the notion of endothelium desquamation as a principal event initiating atherosclerosis (10,11) . More recently, it has become clear that endothelial desquamation is not common and that an intact endothelial cell layer covers developing atherosclerotic lesions. These facts, among others, promoted refinement of the initial hypothesis such that endothelial dysfunction is sufficient to initiate atherogenesis through increased endothelial permeability to atherogenic lipoproteins(12). 2. The response-to-retention hypothesis This hypothesis submits that the lipoprotein retention is the inciting event for atherosclerosis. Within 2 h of injecting LDL into rabbits, arterial retention of LDL and its microaggregates can be observed (13). The underlying mechanisms involved in this process are just now coming to light. It is estimated that 85% of subendothelial lipoprotein delivery is the result of transcytosis, and this process is restricted to particles <70 nm in diameter(14). This size restriction is important as it suggests that lipoprotein lipase activity is needed for triacylgycerol-rich lipoproteins to reach the subendothelial space(15). The retention of lipoproteins within the arterial wall, however, appears tightly linked to components of the extracellular matrix. Apolipoprotein, the single protein associated with LDL, is retained within the arterial wall in close association with arterial proteoglycans (16). This interaction is mediated by specific residues that (17), when mutated, protect experimental animals against the development of atherosclerosis(17,18). In addition to proteoglycan binding, lipolytic and lysosomal enzymes in the extracellular matrix also appear to play a role. For example, lipoprotein lipase enhances the adherence of LDL in vitro (19), and this effect is independent of enzymatic activity(20). Once retained within the arterial wall, LDL can form microaggregates(13,21), as well as lysosomal enzymes such as cathepsin D and lysosomal acid lipase(22). Most importantly, aggregated LDL is avidly taken up by macrophages and smooth muscle cells(23) and thus can support foam cell formation(24). Thus many features of atherosclerosis can be attributed to enhanced retention of LDL within the arterial wall and its association with proteoglycans(6). 3. The oxidative modification hypothesis The oxidative modification hypothesis focuses on the concept that LDL in its native state is not atherogenic. However, LDL modified chemically is readily internalized by macrophages through a so-called "scavenger receptor" pathway(25). Exposure to vascular cells in medium that contains transition metals also results in modification of LDL such that it serves as a ligand for the scavenger receptor pathway(26). It is now clear that one mechanism whereby cells in vitro render LDL a substrate for the scavenger receptor pathway is via oxidation of LDL lipids and the resulting modification of apolipoprotein (27). These observations form the basis for the oxidative modification hypothesis of atherosclerosis , in which LDL traverses the subendothelial space of lesion-prone arterial sites. During this process, LDL lipids are subject to oxidation and, as a consequence, apolipoprotein lysine groups are modified so that the net negative charge of the lipoprotein particle increases(28). This modification of apolipoprotein renders LDL susceptible to macrophage uptake via a number of scavenger receptor pathways producing cholesterol ester-laden foam cells(29). It is this accumulation of foam cells that forms the nidus of a developing atherosclerotic lesion. The process of LDL oxidation is associated with a number of other potentially proatherogenic events. For example, during the initial stages of in vitro LDL oxidation, modification of LDL lipids can occur in the absence of any changes to apolipoprotein. Such modified LDL has been termed "minimally modified LDL" and shown in vitro to induce the synthesis of monocyte chemotactic protein-1 in both smooth muscle and endothelial cells(30,31), resulting in the recruitment of inflammatory cells (32). This particular step appears critical as mice lacking the receptor for monocyte chemotactic protein-1 are resistant to atherosclerosis(33,34). More heavily in vitro oxidized LDL, commonly termed "ox-LDL," is chemotactic for monocytes(35) and T lymphocytes(36), perhaps as the result of lysophosphatidylcholine formed during oxidation(27). Oxidized LDL has also been shown to stimulate the proliferation of smooth muscle cells(37) and to be immunogenic by eliciting the production of autoantibodies (38,39) and the formation of immune complexes that can also facilitate macrophage internalization of LDL(40,41). The recruitment of inflammatory cells may result in the continued oxidation of LDL, setting the stage for catalytic expansion of the atherosclerotic lesion and the full-blown spectrum of atherosclerosis. In summary, the hypotheses of atherosclerosis have each attempted to explain the complex cellular events of atherosclerosis along a common theme. The response-to-injury hypothesis focuses on vascular injury as the inciting event in atherosclerosis with the entire spectrum of the disease representing an attempt to heal an ongoing vascular insult. In contrast, the response-to-retention hypothesis uses lipoprotein-matrix interactions as the critical event in early atherosclerosis, whereas the oxidative modification hypothesis requires oxidation of LDL lipids. Although each hypothesis points to its own critical initiating event, there are many common features among these competing hypotheses. For example, each involves a significant component of inflammation, a known feature of atherosclerosis . Each hypothesis also includes LDL as a central element, an important point as reduction in LDL cholesterol is among the most effective means of treating atherosclerosis. Perhaps the most unique, however, is the oxidative modification hypothesis as it alone proposes a particular importance of oxidative events and redox reactions in the genesis of vascular disease (6). 1.2 Oxidative Stress Oxidative stress which can be defined as an imbalance develops between the production of ROS and the efficacy of the cell's antioxidant defense, leading to an altered redox status which can contribute to endothelial dysfunction and/or cell death(42,43). 1.2.1 Free radicals Free radicals can be defined as molecules or molecular fragments containing one or more unpaired electrons. The presence of unpaired electrons usually confers a considerable degree of reactivity upon a free radical. Those radicals, derived from oxygen, represent the most important class of such species generated in living systems (44). These free radicals are highly reactive, unstable molecules react with (oxidize) various cellular components including DNA, proteins, lipids / fatty acids and advanced glycation end products (e.g. carbonyls) which lead to DNA damage, mitochondrial malfunction, cell membrane damage and eventually cell death (apoptosis - which is the term for programmed cell death)(45). Free radicals are generally reactive oxygen species (ROS) or reactive nitrogen species(RNS), and have consequences for cells as a result of one of three factors: (i) an increase in oxidant generation, (ii) a decrease in antioxidant protection, or (iii) a failure to repair oxidative damage (46). Examples of free radicals (oxidizing molecules) are hydrogen peroxide, hydroxyl radical, nitric oxide, peroxynitrite, single oxygen, superoxide anion and peroxyl radical(45). 1.2.2 Oxidative stress and reactive oxygen species In the vascular system, the formation of ROS from endothelial cells, smooth muscle cells (SMCs) and macrophages seems to be of major relevance in atherogenesis in part due to their reaction with nitric oxide (NO). NO, perhaps the most important endothelium-derived vasorelaxing factor, is scavenged by ROS. NO reacts with ROS to yield peroxynitrite, which can rearrange to form nitrate and the highly reactive OH radical and the last is toxic to tissues and cells. Important pathophysiological consequences of enhanced ROS formation in the vascular system are: (i) attenuation of endothelium-dependent dilation, resulting in disturbed organ perfusion and systemic hypertension, (ii) induction of cellular damage and inflammation, (iii) induction of apoptosis, and (iv) initiation of several intracellular signaling processes (47). 1.2.3 Sources and reactions of ROS and RNS ROS can be produced from both endogenous and exogenous substances. Potential endogenous sources include mitochondria, cytochrome P450 metabolism, peroxisomes, and inflammatory cell activation (48). Mitochondria have long been known to generate significant quantities of hydrogen peroxide. The hydrogen peroxide molecule does not contain an unpaired electron and thus is not a radical species. Under physiological conditions, the production of hydrogen peroxide is estimated to account for about ∼2% of the total oxygen uptake by the organism. After the determination of the ratios of the mitochondrial generation of superoxide to that of hydrogen peroxide, the former was considered as the stoichiometric precursor for the latter. Ubisemiquinone has been proposed as the main reductant of oxygen in mitochondrial membranes(48). Mitochondria generate approximately 2–3 nmol of superoxide/min per mg of protein, the ubiquitous presence of which indicates that it is the most important physiological source of this radical in living organisms(48). Since mitochondria are the major site of free radical generation, they are highly enriched with antioxidants including GSH and enzymes, such as superoxide dismutase (SOD) and glutathione peroxidase (GPx), which are present on both sides of their membranes in order to minimise oxidative stress in the organelle(49). Superoxide radicals formed on both sides of mitochondrial inner membranes are efficiently detoxified initially to hydrogen peroxide and then to water by Cu, Zn-SOD (SOD1, localised in the intermembrane space) and Mn- SOD (SOD2, localised in the matrix). Besides mitochondria, there are other cellular sources of superoxide radical, for example xanthine oxidase (XO), a highly versatile enzyme that is widely distributed among species (from bacteria to man) and within the various tissues of mammals(50). Xanthine oxidase is an important source of oxygen-free radicals. It is a member of a group of enzymes known as molybdenum iron–sulphur flavin hydroxylases and catalyzes the hydroxylation of purines. In particular, XO catalyzes the reaction of hypoxanthine to xanthine and xanthine to uric acid. In both steps, molecular oxygen is reduced, forming the superoxide anion in the first step and hydrogen peroxide in the second(44). Additional endogenous sources of cellular reactive oxygen species are neutrophils, eosinophils and macrophages. Activated macrophages initiate an increase in oxygen uptake that gives rise to a variety of reactive oxygen species, including superoxide anion, nitric oxide and hydrogen peroxide(51). Cytochrome P450 has also been proposed as a source of reactive oxygen species, through the induction of cytochrome(44) . 1.2.4 Antioxidants Under normal conditions, numerous cellular antioxidant systems exist to defend against oxidant stress and maintain the redox balance of the cell. ROS are cleared from the cell by enzymatic systems including superoxide dismutases (SODs), catalase, and glutathione peroxidase, or the nonenzymatic system including alpha-tocopherol (vitamin E), ascorbic acid (vitamin C), glutathione, and uric acid. SODs convert two superoxide anions into one molecule of hydrogen peroxide and oxygen, and catalase catalyzes the decomposition of hydrogen peroxide into water and oxygen, leading to the removal of superoxide anion. Glutathione peroxidase plays an important role as defense mechanism in mammals, birds and fish against oxidative damage by catalyzing the reduction of a variety of hydroperoxides, using glutathione as the reducing substrate. For example, glutathione peroxidase reduces hydrogen peroxide to water by oxidizing glutathione (GSH) to its oxidized form (GSSG), and the reduction of the oxidized form of glutathione (GSSG) is then catalyzed by glutathione reductase. Through the glutathione redox cycle, ROS is removed from cells. In addition to its role as a substrate in GSH redox cycle, glutathione, as well as uric acid, also act as a direct endogenous scavenger of hydroxyl radicals. Ascorbic acid scavenges free radicals and reduces them into hydrogen peroxide. Hydrogen peroxide can be further catalyzed by catalase to form water and oxygen. Alphatocopherol can transfer a hydrogen atom with a single electron to free radicals, thus removing the radicals before they can interact with cell membrane proteins or generate lipid peroxidation(52). 1.2.5 Oxidative stress in atherosclerosis Oxidative stress has been identified throughout the process of atherogenesis, beginning at the early stage when endothelial dysfunction is barely apparent(53). As the process of atherogenesis proceeds, inflammatory cells, as well as other constituents of the atherosclerotic plaque release large amounts of ROS, which further facilitate atherogenesis. In general, increased production of ROS may affect four fundamental mechanisms that contribute to atherogenesis: oxidation of LDL, endothelial cell dysfunction, vascular SMCs, growth and monocytes migration(54). A number of studies suggest that ROS oxidize lipids and that the oxidatively modified LDL is a more potent proatherosclerotic mediator than the native unmodified LDL(55). The suggestion is based on the observations that high plasma levels of ox-LDL are present in patients with atherosclerosis and that antibody to ox-LDL is detected in plasma of most patients with atherosclerosis(56). Strong evidence in favor of a proatherosclerotic role for ox-LDL comes from a number of studies demonstrating the noxious effects of ox-LDL on various components of the arterial wall. For example, ox-LDL causes activation of the endothelial cells lining the arterial wall, resulting in the expression of several adhesion molecules that facilitate the adhesion of monocytes/macrophages (57). oxLDL also activates inflammatory cells and facilitates the release of a number of growth factors from monocytes/macrophages(58,59). Vascular SMCs exhibit intense proliferation when exposed to ox-LDL. ox-LDL enhances the formation of matrix metalloproteinases (MMPs) in vascular endothelial cells and fibroblasts(60) thus setting the stage wherein oxidative stress leads to rupture of a soft plaque. In addition, ox-LDL upregulates the expression of its endothelial receptor LOX-1 and other scavenger receptors mainly expressed on macrophages/monocytes. The increased expression of these receptors is responsible for the uptake of ox-LDL and the formation of foam cells, which is an early step in atherogenesis(45). 1.3 Endothelium dysfunction 1.3.1 Endothelial dysfunction as an initial step of atherosclerosis The endothelium is a flat monolayer of cells that cover vascular lumina throughout the body. In adult, the number of endothelial cells is approximately one trillion. The total surface area of the endothelium is as large as six tennis courts and the weight of whole cells is larger than that of the liver. In other words, the endothelium is the largest organ in the body(61). Endothelial cells play diverse biological roles, such as maintaining vascular tone and structure, regulating intravascular hemostasis and permeability, and cell adhesion and migration(62). Recent progress in vascular biology has revealed that the endothelium releases a large number of vasoactive substances. These substances are divided into two classes: endothelium-derived relaxing factors (EDRFs), and endothelium-derived contracting factors (EDCFs). It has been shown that EDRFs such as nitric oxide (NO). Endothelial Dysfunction as an Initial Step for Atherosclerosis which caused by imbalance between EDRFs and EDCFs precedes and promotes atherosclerosis by several mechanisms, such as adhesion of monocytes and platelets;and increase in vascular permeability, and proliferation and migration of smooth muscle cells. These processes result in plaque formation and intimal thickening, whereas EDCF such as endothelin have opposite effect and participate in the progression of cardiovascular diseases. The exposure to coronary risk factors decreases the bioactivity of EDRFs and increases the release of EDCFs(63,64). Thus, coronary risk factors impair the bioactivity of NO derived from endothelial cells and the impaired bioactivity of NO exacerbates atherosclerosis(65,66). Furthermore, it has been shown that atherosclerosis by itself decreases biological NO activity, leading to a vicious cycle(65). 1.3.2 Oxidative stress and endothelial dysfunction Oxidative stress induced by reactive oxygen species impair the biological activities of endothelium-derived NO through several mechanisms. Endothelium-dependent vasodilation was lost at basal level and was restored by anti-oxidant ascorbic acid in patients with mitochondrial diseases, suggesting that oxidative stress may be involved in premature cardiovascular diseases and anti-oxidant may become a therapeutic tool in mitochondrial diseases. However, it remains unknown whether the circulating form of oxidized LDL impairs endothelial function (67). 1.3.3 Antioxidant properties of nitric oxide in atherosclerosis Nitric oxide has many physiological actions that can be interpreted to be potentially antiatherosclerotic(68,69). It inhibits 1) platelet aggregation and adherence to endothelial cells, 2) monocyte adherence to endothelial cells, 3) the expression of the monocyte chemoattractant protein, 4) vascular smooth muscle cell migration and proliferation and 5) the in vivo intimal proliferative response to ballon injury. Nitric oxide reduces oxidant stress in the vascular wall, which in turn may lower the rate of LDL oxidation and the expression of redox-sensitive genes that contribute to atherogenesis. In fact, vascular NO either suppresses the expression of adhesion molecules by endothelial cells or the generation of products that are chemotactic for monocytes such as oxidized LDL. There is accumulating evidence that the salutary effects of NO are diminished in atherosclerotic vessels due to its reactions with reactive oxygen species. In particular, the reaction of NO with O2•-, as well as its reaction with LO. and LOO. to inhibit lipid oxidation, suggests that NO can both enhance and inhibit lipoprotein oxidation in the vessel wall. The removal of NO from the vascular compartment by its rapid reactions with these free radical species will concomitantly lower its steady state concentration, and hence increasing platelet and inflammatory cell adhesion to the vessel wall and impairing endothelial-dependent mechanisms of relaxation(70). Nitric oxide has been observed to play a critical role in regulating lipid oxidation induced by reactive oxygen and nitrogen species (71,72). Nitric oxide and mechanisms underlying impaired vasomotor responses in atherosclerosis. Endothelium plays an important role in maintaining vascular integrity by the synthesis and release of vasoactive substances such as NO. The changes that occur during atherosclerosis include the loss of the control of vascular tone, a NO-dependent event. The mechanisms accounting for endothelial dysfunction in hypercholesterolemia have not been completely elucidated, but may be explained by decreased bioavailability of NO due to either decreased expression of the eNOS, decreased substrate availability, presence of an endogenous eNOS inhibitor or increased NO degradation by reactive oxygen-and nitrogen species(73). 1.4 Treatment of Atherosclerosis The treatment of atherosclerosis relies heavily on the reduction of risk factors particularly elevated serum lipids level, hypertension, diabetes and cigarette smoking. The incidence of cardiac and cerebral infarction can be diminished considerably by controlling risk factors. Lifestyle factors are also of greatest importance in the treatment of atherosclerosis(74,75). 1.4.1 Lifestyle change The most common treatments focus on lifestyle changes to reduce cholesterol and other problems that contribute to atherosclerosis. Dietary modifications usually incorporate eating foods that are low in saturated fats, cholesterol, sugar, and animal proteins. Foods high in fiber, such as fresh fruits and vegetables, and whole grains, are encouraged. By consuming fruits and vegetables, the person also consumes helpful dietary antioxidants, such as carotenoids found in vegetable pigments, and bioflavenoids in fruit pigments. Liberal use of onions and garlic is recommended, as well as eating fish, especially cold-water fish, such as salmon. Smoking, alcohol, and coffee are to be avoided, and exercise is strongly recommended(76). 1.4.2 Medications Medications commonly prescribed in the treatment of atherosclerosis include anti-hypertensives and cholesterol reducing drugs(77). 1.Anti-hypertensives While anti-hypertensives do not control the process of atherosclerosis, they are successful in controlling one of the primary side effects. These drugs include calcium channel blockers, ACE inhibitors, and angiotensin receptor blockers(78). 2. Cholesterol reducing drugs Keeping serum cholesterol level in the normal range not only helps prevent heart attacks and strokes but may also prevent the progression of atherosclerosis(79). These drugs include statins, bile-acid–binding resins, nicotinic acid, fibrates and cholesterol absorption inhibitors (ezetimibe) . 1.4.3 Therapeutic approach It is hoped that understanding the biology of the endothelium in atherosclerosis will lead to therapies that will retard its progression, and reduce the incidence or consequences of acute complications. At present, insights from endothelial biology have provided an explanation of the mechanism of action of existing therapies (NO donors, aspirin), but new treatments have been slow to emerge. Endothelin antagonists have been developed for the treatment of pulmonary hypertension. However their usefulness in the treatment of human atherosclerosis has been hampered by toxicity. L-arginine supplementation can retard the progression of atherosclerosis in animal models . Short-term studies suggest that Larginine supplementation can restore conduit and resistance vessel endotheliumdependent dilatation in humans with risk factors for atherosclerosis. It is possible that modulation of the BH4 pathway in humans may be a future therapeutic target. Whether measures to preserve endothelial function may reduce the incidence of acute vascular events, or limit tissue damage during ischaemia-reperfusion injury, remains to be seen . Sildenafil, an inhibitor of cGMP breakdown by phosphodiesterase V, has been a therapeutic advance in the treatment erectile dysfunction caused by atherosclerotic disease, but its potential for the treatment of the systemic vasculature is uncertain. Therefore our study is undertaken to assess the effect of phosphodiesterae inhibitor on systemic vasculature in experimental model of atherosclerosis(approach)(80). 1.5 Phosphodiesterase Inhibitors Phosphodiesterases (PDEs) are a superfamily of enzymes involved in regulating intracellular signaling by cleaving cyclic adenosine monophosphate (cAMP) or cyclic guanine monophosphate (cGMP) to their corresponding’ nucleotides, and thus rendering them inactive. Cyclic AMP is a ubiquitous second messenger that controls cell functions such as growth, differentiation, survival and inflammation by affecting protein kinase activators (PKA) in response to Gs or Gi coupled receptor activation. PDEs act as a critical feedback mechanism in the cAMP cellular signaling cascade. There are at least 11 PDE families recognized in mammals that have been characterized with respect to biochemical properties, expression, and their affects on other proteins or pathways. PDEs have become novel therapeutic drug targets. They share common catalytic sequences but they differ in substrate specificity, tissue distribution and expression, sensitivity to inhibitors and mode of regulation(81). 1.5.1 Sildenafil Sildenafil belongs to a class of compounds called selective PDE-5inhibitors(82). Sildenafil was synthesized by a group of pharmaceutical chemists working at Pfizer's . It was initially studied for use in hypertension and angina pectoris. The first clinical trials, suggested that the drug had little effect on angina, but that it could induce marked penile erections(83). Pfizer therefore decided to market it for erectile dysfunction by the US Food and Drug Administration on March 27, 1998, becoming the first oral treatment approved to treat erectile dysfunction in the United States, and offered for sale in the United States later that year(84). 1.5.1.1 Mechanism of action and pharmacological properties Sildenafil is a potent inhibitor of PDE-5 which is the predominant isozyme that metabolizes cGMP in the corpora cavernosa of the penis. cGMP is the second messenger of NO and a principal mediator of smooth muscle relaxation and vasodilatation in the penis (82). Sildenafil is a potent inhibitor of PDE-5, with favorable selectivity (>1000-fold) for human PDE-5 over human PDE2 (in the adrenal cortex), PDE3 (in smooth muscles, platelets, and cardiac tissue),and PDE4 (in the brain and lung lymphocytes) and moderate selectivity (>80 fold ) over PDE1 (in the brain, kidney, and smooth muscle). Sildenafil is only 10-fold as potent for PDE5 as for PDE6(82). In addition, sildenafil possess direct muscle relaxant potential possibly via inhibiting Ca(2+) influx through both receptor-operated and voltage dependent Ca(2+) channels(85) and induce vasorelaxation that is associated with increases in the phosphorylation of heat shock related protein 20 (HSP20) (86). 1.5.1.2 Effect on hemodynamics The data from hemodynamic studies would suggest that sildenafil is a modest vasodilator with effects on both the venous and arterial tree (87). It possesses vasodilatory properties, which result in mild, insignificant decreases in BP when taken alone (82). Sildenafil improves microcirculation in patients with Raynaud’s phenomenon(88,89).. During exercise and recovery, sildenafil does not cause clinically significant alterations in hemodynamic parameters in men with CAD(90). Sildenafil produces a transient modest reduction in systolic and diastolic blood pressures (BP). No significant effects are observed on heart rate (HR) (82), while systemic BP did not change after sildenafil administration (2 mg/kg), and the blood flow in a normal coronary artery increased(91). Sildenafil if used regularly may be responsible for small yet significant decrease in BP in diabetic hypertensive patients with erectile dysfunction along with improvement in their sexual dysfunction (92) . Sildenafil may be useful in treating patients with HF, PH, and CAD(93). Also sildenafil has no direct effect on cardiac repolarization (QT interval)(94,95). The combination of sildenafil with any agent acting as a NO donor is contraindicated due to life-threatening hypotension and the increment in ventricular tachycardia/ventricular fibrillation (VT/VF) vulnerability in the normal right ventricles (RV)(96). It also causes a marked increase in sympathetic activation, evident both at rest and during stressful stimuli (97). It was found that sildenafil blunts systolic responses to ßadrenergic stimulation and has potent effects on hearts stimulated by ßadrenergic or pressure overloads (98). Sildenafil does not worsen exercise capacity and exercise-induced myocardial ischemia (99), while it reduces cardiovascular remodeling associated with hypertensive cardiomyopathy (100) . Sildenafil has a cardioprotective effect and anti-ischemic effect (101). Sildenafil inhibits platelet activation in patients with CAD (102). 1.5.1.3 Pharmacokinetics and metabolism Sildenafil is rapidly absorbed orally, with absolute bioavailability of 40%. Sildenafil is highly selective for the cGMPhydrolyzing isoform 5 with a half-maximal inhibition (IC50) of PDE-5 activity at a concentration of 3.5 nmol/L, followed by IC50 values of 34 to 38 nmol/L for PDE 6 (in the retina) and 280 nmol/L for PDE 1. The cAMPhydrolyzing PDE 3 and 4 and the cAMP- and cGMP-hydrolyzing isoform 2, as well as PDE 7 to 11,are inhibited by sildenafil with an IC50 of >2600 nmol/L(82). Plasma concentrations peak within 30 to 120 minutes of oral dosing in the fasted state. Sildenafil is primarily metabolized by the cytochrome(cyt.)P450 3A4 hepatic microsomal isoenzymes,which convert it to an active N-desmethyl metabolite. Sildenafil and its active metabolite are both highly bound to plasma proteins (96%), and their terminal half-lives are 4 hours each. sildenafil is excreted as metabolites predominantly in the feces. Plasma levels of sildenafil are increased in patients aged >65 years and in patients with hepatic impairment, severe renal impairment, and concomitant use of erythromycin , cimetidine, ketoconazole, fluvoxamine and indinavir and grapefruit juice increases sildenafil bioavailability(82). 1.5.1.4 Adverse effects Sildenafil has these adverse effects: headache, flushing, rhinitis , nasal obstruction ,dizziness, hypotension, postural hypotension, dyspepsia ,burning sensation, increased perception of light, and blurred vision , myalgias, changes in serum creatine kinase, priapism,a transient prolongation of the BT, intracerebral hemorrhage, gastric variceal bleeding, epistaxis, and hemorrhoidal bleeding, tonic-clonic seizures, transient ischemic attacks, strokes, and transient global amnesia,.hepatotoxicity, atrial fibrillation, migraine, stroke, gallbladder disorders, lichenoid drug eruption ,toxic epidermal necrolysis, MI, vestibular neuritis-like symptoms(82). 1.5.1.5 Drug interaction Vasodilator actions of nitrates are amplified with concomitant use of sildenafil within the first 24 hours(82,103). Sildenafil is an inhibitor of the cyt. P450 2C9 metabolic pathway. Therefore, administration of sildenafil could result in a significant increase in the plasma concentrations of other drugs metabolized through this pathway. sildenafil is predominantly metabolized by both the P450 2C9 pathway and the P450 3A4 pathway. Thus , cimetidine ,ciprofloxacin , clarithromycin ,erythromycin, itraconazole, nefazodone, diltiazem, ritonavir and grapefruit juice increases the plasma concentrations of sildenafil .Sildenafil should be used with caution in patients who take alphablockers,and nicorandil(103). 1.5.2 Vardenafil Vardenafil is the second oral selective PDE-5 inhibitor for erectile dysfunctin with the highest potency and minimal inhibition of other PDEs(104). It was licensed in Europe in 2003 and in the USA in late 2003 (105). Vardenafil is a PDE5 inhibitor used for treating impotence (erectile dysfunction) that is sold under the trade name Levitra (Bayer AG, GSK, and SP)( 106). Vardenafil's indications and contra-indications are the same as with other PDE5 inhibitors. It is closely related in function to sildenafil (Viagra) and tadalafil (Cialis). Structurally, the difference between the vardenafil molecule and sildenafil is a nitrogen atom's position and the change of sildenafil's piperazine ring methyl group to an ethyl group(106). 1.5.2.1 Mechanism of action and pharmacological properties The mechanism of action and pharmacological properties of sildenafil and vardenafil are comprehensively alike. 1.5.2.2 Hemodynamic effects Recent studies suggest that there are pharmacological differences between the three available PDE5- inhibitors. Gofrani et al. compared the short-term haemodynamic effects of sildenafil, vardenafil and tadalafil in a well-defined population suffering from chronic pulmonary arterial hypertension. Results showed that vardenafil lacked selectivity for the pulmonary circulation, while only sildenafil improved arterial oxygenation. In another study, Teixeira et al. demonstrated that vardenafil, but not sildenafil or tadalafil, affects Ca2+ handling in the rat aorta besides increasing cGMP levels(107). The ability to exercise among patients with CAD is not impaired with vardenafil treatment (108).Vardenafil demonstrats clinically significant differences (fainting) with respect to sildenafil only during the first doses(109). Vardenafil produces a slight hypotensive effect and a minor compensatory increase in heart rate(110). While, unlike sildenafil, vardenafil has been associated with a slight prolongation of the QT interval (111) . However, the co-administration of vardenafil with tamsulosin is not associated with clinical significant hypotension (112). The study by Deibert et al., showed that vardenafil (another phosphodiesterase type-5 inhibitor) was found to lower portal pressure in four of five patients with Child A cirrhosis(113). In investigating the effects of vardenafil, on myocardial and endothelial functions, it was conclude that the myocardial contractility parameters showed no significant changes when compared to that at baseline in addition to an initial drop of blood pressure up to 15—20%(114). Vardenafil induces a protective effect against ischemia/reperfusion injury(115), and specifically relaxes coronary resistance vessels(116). However, Thadani et al. ,studied men with reproducible angina on exercise and showed that vardenafil 10 mg did not change the total exercise time or time to angina compared with placebo and actually increased the time to ischemic STsegment changes(117). While Vardenafil has no effect on the pharmacodynamics of warfarin (prothrombin-time),and aspirin (bleeding time)(118). 1.5.2.3 Pharmacokinetics Vardenafil needs lower absolute concentrations to inhibit PDE-5. However, relative selectivity for PDE-5 is similar to that of sildenafil(119). It shares the molecular structure of sildenafil(120). The time to peak serum concentration of vardenafil is 0.75 hour comparable to that of sildenafil (1.16 hours).It is metabolized by the liver and excreted mainly in the feces with an excretion half-life of 4.7 hours, only longer than that of sildenafil(approximately 4 hours) and rapid onset of action. It can be taken after eating a moderately fatty meal.Its potency was approximately 25 times greater than that of sildenafil and 48 times greater than that of tadalafil while a high-fat meal may alter Cmax slightly and delay the absorption up to 1 hour, a moderate-fat meal has no effect on vardenafil pharmacokinetics. Ritonavir can affect its hepatic metabolism(121). 1.5.2.4 Side effects Vardenafil has these adverse effects: headaches, flushing, rhinitis, dyspepsia, epileptic seizures, atrial fibrillation, with less visual than sildenafil changes, and urticaria(122,123). 1.5.2.5 Drug interaction Vardenafil may potentiate the hypotensive effects of nitrates(123); its lack of interaction at 24 hours(120). Vardenafil increases the hypotensive effect of α-adrenergic blocking drugs except tamsulosin(124). Briganti et al. (2005 ), (121) showed that ritonavir, indinavir, erythromycin, itraconazole, and ketoconazole reduce vardenafil clearance. 1.6 Doppler Ultrasound Technique Duplex and triplex U.S. techniques are non-invasive diagnostic techniques that obtain flow velocity data and used as an alternative to invasive intra-arterial angiography which is associated with local and systemic complications, and to acquire direct physiologic information about affected arteries. Duplex U.S. combines a B-mode ultrasound image with a pulse wave Doppler unit; whereas triplex U.S combines a B-mode ultrasound image with a color flow unit and a pulse wave Doppler unit(125). The advantages of duplex and triplex U.S. include absence of complications, relatively low costs, and widespread availability(126). These techniques provide an accurate evaluation of hemodynamically significant occlusive vascular lesions. They are used in the evaluation of the extracranial carotid arteries, iliofernoral arteries, abdominal arteries and hemodialysis shunts. These systems obtain information regarding blood flow velocity, lumen area, plaque composition and ulceration, and hemodynamic turbulence(127). With duplex and triplex U.S. techniques, the severity of a stenosis can be determined by using peak systolic velocity (PSV) measurements in arteries with reduced luminal diameter, the PSV ratio at the site of the stenosis and the adjacent normal artery, the end-diastolic velocity and other less firmly established criteria(128). 1.6.1 Doppler parameters and doppler indices Peak systolic velocity (PSV) Peak systolic velocity is a measure of the maximum velocity of blood flow during systole. Using Doppler principles, as an artery narrows, the velocity of blood flow increases(129). So the peak systolic velocity is used as a criteria for diagnosis of arterial stenosis, e.g. the PSV in the renal artery is less than 180 cm/s in normal adult human, an increase in peak systolic flow velocity greater than 180 cm/s indicates a renal artery stenosis of more than 50% diameter reduction(130). End diastolic velocity (EDV) End diastolic velocity is a measure of the velocity of blood flow at the end of diastole. End diastolic velocity is a measure of resistance in the distal arteries, where high resistance in the distal vessels produce low diastolic flow in the supplying artery(131). Mean velocity Mean velocity is the average blood flow velocity during the cardiac cycle. Sometimes mean velocity is used together with the vessel crosssectional area to calculate blood flow rate. However, it is difficult to measure mean velocity accurately and there are several other problems associated with calculating flow rate(132). Resistive index (RI) and Pulsatality index (PI) Resistive index (RI) is a pulsed-wave Doppler index used for measurements of downstream resistance in arteries (high resistance in the distal vessels produce low diastolic flow in the supplying arteries and result in a high value for this index and vise versa)(131). Pulsatality index ( PI) is also a pulsed-wave Doppler index designed to quantify the pulsatality or oscillations of the blood flow waveforms(131). Originally both PI and RI were introduced to detect peripheral vascular diseases, but they are rarely used in these conditions(133). However, if the indices are measured in renal arteries, they are reported as reliable measurements of down-stream renal resistance(134). An increasing amount of information is available about the usefulness of PI and RI in the investigation and monitoring of renal artery stenosis(135,136). Ohta et al. (2005) reported that increased RI and PI of the renal arteries is associated with arterial stiffness and reflects the severity of systemic atherosclerosis(137). RI and PI are calculated via soft wave programs built in all recent Doppler machines. Renal artery-to-aortic PSV ratio (RAR) Renal-aortic ratio is the ratio of the PSV in the renal artery to the PSV in the aorta . This Doppler index is used as a criteria for diagnosis of renal artery stenosis. In adult human the normal value of renal-aortic ratio is less than 3.5, RAR value equal to or more than 3.5 indicates a 60% renal artery stenosis or more(129). 1.7 Blood Flow Rate and Blood Flow Velocity 1.7.1 Definition of flow rate and flow velocity When considering flow in a system of tubes, it is important to distinguish between velocity which is displacement per unit time (e.g. cm/s) and flow rate which is volume per unit time ( e.g. ml/s)(138). 1.7.2 Factors affecting on blood flow and blood flow velocity There is a linear relationship between blood flow rate and peak blood flow velocity in fixed diameter vessels with laminar flow. So with the exception of the effect of radius ( diameter ) of blood vessel on blood flow rate, any other factor which increase or decrease blood flow rate will increases or decreases the blood flow velocity(139). The factors influencing on blood flow rate are explained by poiseuilles law : F = P r 4 / 8 l. In which F is the rate of blood flow, P is the pressure difference between the ends of the vessel, r is the radius of the vessel, l is the length of the vessel and is the viscosity of the blood(140). So factors affecting on blood flow are: 1.Vessel radius The blood flow rate is directly proportional to the fourth power of the radius of the vessel which demonstrates the diameter of blood vessel (which is equal to twice the radius). So as the vessel radius is increased the blood flow rate increased. But there is inverse relationship between vessel radius and blood flow velocity(138). 2.Viscosity : As the blood viscosity increased the blood flow rate and blood flow velocity are decreased, so the factors affecting on blood viscosity will affect on blood flow rate and velocity. The most important factor is the hematocrite. Gruber et al. (1999), and Rosenkrantz et al. (1984) found that blood flow rate and velocity are increased in case of anemia and decreased in case of polycythemia and dehydration (141,142). Blood viscosity increases when blood temperature decreases. However, the increase in viscosity is observed mostly at temperature below 15°C. So when blood temperature is reduced, the blood flow rate and velocity are reduced(143). 3. Pressure gradient P As the pressure gradient increased, the blood flow rate is increased. Factors affecting on pressure gradient are cardiac output, peripheral resistance, and vessel compliance(144). 1.7.3 Effect of atherosclerosis on blood flow velocity Healthy blood vessels are elastic and flexible (compliant) (145). Atherosclerosis is a form of the disease that causes blood vessels to become thick and stiff because of fatty plaques that develop inside of arterial walls(146). When atherosclerosis first begins, the fatty plaque grows and begins to protrude into the blood vessel wall and restricts the amount of blood that can flow past it. This type of blockage is called a stenosis. The newly blocked vessel now has the same amount of blood trying to flow through a smaller space. This causes an increase in the velocity of the blood flow at the point where the stenosis is located(147,148). 1.7.4 Aortic blood flow The normal Doppler wave forms of the aorta varies with the location. In the upper aorta, there is a narrow well defined systolic complex with forward flow during diastole. Below the renal arteries the diastolic flow is much reduced and above the bifurcation it is absent or reversed(149). The main abnormalities affecting the aorta are atheroma and aneurysm. Atheroma can affect the aorta and produce stenosis or occlusion. In such cases velocity ratios taken from above and at the stenosis can be used to assess the degree of hemodynamic compromise, if there is significant increase in the blood flow velocity and velocity ratio by a factor of two or more, it would indicate a sever aortic stenosis(150). 1.7.5 Renal artery blood flow To a large extent, the excretory and regulatory processes of the kidney depend on the blood supply to the kidney. Therefore, it is not surprising that it receives the highest blood flow per gram of organ weight in the body. The combined blood flow through both kidneys is about 1100 ml/min which is about 22% of the total cardiac output. Thus, a very substantial portion of the total cardiac output flows through the kidneys(151). The normal Doppler wave forms of the renal arteries in normal adult human are characterized by a rapid systolic upstroke which is occasionally followed by a secondary slower rise to peak systole and there is subsequently a gradual diastolic decay but with persistent forward flow in diastole. In normal adult human, the peak systolic velocity of the normal renal artery is less than 180 cm/s(152). 1.7.6 Intra-renal arteries blood flow The renal arteries branches into segmental arteries which subdivide into interlobar arteries. Each interlobar artery penetrates the kidney through a column of Bertin, before reaching the cortical surface divides into arcshaped arcuate arteries. The normal Doppler wave forms of segmental arteries in normal adult human at rest are pulsatile, diphasic, and characterized by sharp rapid upstroke, having double systolic peak, and of high diastolic flow(153). In normal adult human at supine position, the interlobar intra-renal arteries peak systolic velocity and end diastolic velocity are in the range of 32-58 cm/s and 13-18 cm/s respectively(154). 1.8 Aim of the Study This study was undertaken to evaluate the effects of sildenafil and vardenafil on atherosclerosis progression. 2.1 Materials The chemicals, drug and instruments used in the present study with their supplier listed in the following tables Table (1): list of chemicals pharmaceuticals, reagent and their supplier Pharmaceuticals and reagents Supplier Sildenafil pfizer,USA, Batch No 89R001E Vardenafil Bayer AG,Germany. Batch no.NBRYO6. pure cholesterol powder ( C27H46O) M. wt = 386.67 BDH Chemicals Ltd Poole England. Total cholesterol ,Triglyceride and HDL Reagents BIOLABO SA ,02160MAIZYFRANCE Reduced Glutathione (GSH) Biochemical Trichloroacetic acid (TCA) Merk, Germany 5,5-dithiobis(2-nitrobenzoic acid) DTNB Sigma Ethylene Diamineteteraacetic acid Disodium(EDTA) BDH,U.K Trishydroxymethylene Merk, Germany Absolute Methanol Fluka Thiobarbaturic acid (TBA) Merk Co.Ltd Table (2): Instrument and their supplier Equipment Company Sensitive electrical balance Sartorius /Germany pH meter Inolab /Germany spectrophotometer Shimadzu UV-1650(UV-visible)/japan spectrophotometer Apple UK Vortex (super mixer) LAB-Line /USA Centrifuge Hettich/Germany Water bath Memmert /Germany 2.2 Animals and study design 24 local domestic rabbits were used in this study. Their weight ranged between 1.4-1.7kg, they were housed in the animal house of Al Kufa College of medicine. They were kept in cages under a 12-h light:12-h dark cycle at room temperature 25°C, and humidity was kept at 60–65%. , and they were allowed to drink tap water and given standard chow diet at libitum. During 1 wk of adaptation; alfalfa and other grasses or vegetables were given but with small amounts until the end of this week when they were prevented completely to avoid their influence on hypercholesterolemic atherosclerosis protocol. After the 1st week of acclimatization the rabbits were randomized into four groups as follow: i. Normal control group: This group consists of 6 rabbits; all rabbits of this group were kept on standard chow diet and tap water throughout the duration of study (12 weeks). ii. High cholesterol diet control group: (induced untreated group): This group consists of 6 rabbits; all rabbits of this group were kept on atherogenic diet (a 2% cholesterol-enriched diet) and tap water throughout the duration of study (12 weeks). iii. Sildenafil treated group: This group consists of 6 rabbits. All rabbits in this group were kept on atherogenic diet ( 2% cholesterol enriched diet) and tap water for 6weeks, then they were treated with sildenafil 5 mg./kg. body weight / day orally(155) in a single dose at evening for the next 6 weeks . The treatment continued together with the atherogenic diet. iv. Vardenafil treated group: This group consists of 6 rabbits. All rabbits in this group were kept on atherogenic diet ( 2% cholesterol enriched diet) and tap water for 6weeks. Then they were treated with vardenafil 18 mg./kg. body weight / day orally(156) in a single dose at evening for the next 6 weeks. Also the treatment continued together with the atherogenic diet. The standard diet for rabbits consisted of 10% wheat, 40% grass powder, 12% soybean cake, 20% corn, 10% wheat bran, 3% fish flour, 1% salt, 3% bone meal, and 1% multivitamins (percent by weight)(154). The high-cholesterol experimental diet consisted of 2% cholesterol was made by 20gm of powder cholesterol for each 1000gm of standard rabbit diet. The food intake was recorded every day. The rabbit weigh weekly to ensure proper food consumption among all groups. The experimental induction of atherosclerosis were conducted in accordance with Meena Narayanasawamy, Kenneth C. Wright, Krishna Kandarpa (2000)(158) that use 2% cholesterol for 12 week as model of hypercholesterolemic atherosclerosis in rabbit. 2.3 Drugs 2.3.1 sildenafil Sildenafil used in dose of 5 mg / kg. A tablet dissolved in 15 cc DW ,in concentration of 5 mg/ 1.5 cc and the dose given according to body weight orally in a single dose at morning .(Sildenafil solubility in D.W. is 3.5 mg/1 cc)(156). 2.3.2 vardenafil Vardenafil used in dose of 18 mg/kg has been used .A tablet dispersed in tap water and the dose given according to the body weight by oral gavages(159). 2.4 Preparation of sample 2.4.1 Blood sampling From each rabbit about 3 ml of blood was collected from the central ear artery without user heparin after an overnight fasting. The blood sampling was done firstly at the start of the study, i.e. at zero time, 6 weeks of the induction period, i.e. at the start of treatment with sildenafil and vardenafil, and then every week of the treatment course. The blood samples were allowed to clot at 37 C and centrifuged at 3000 rpm for 15 min. , Sera were removed, and analyzed for determination of serum total cholesterol, triglycerides, HDL- C and oxidative stress parameter (MDA,GSH). 2.4.2 Tissue sampling At the end of the protocol (12 weeks on their respective diets), rabbits were sacrificed with high dose of Phenobarbital sodium (200mg, intravenously). The killed rabbits were dissected through the chest wall to make the aorta accessible for resection. The aortic arch was exteriorized, cleaned of adherent fat and connective tissue excised. All specimens were immediately fixed in 10% formaldehyde solution. After fixation they were processed in usual manner. The sections were examined by microscope under magnification power of (×10 and ×40) then the histological changes were determined according to American Heart Association classification of atherosclerosis phases(160,161) which divides atherosclerotic lesion into different degree and phases as follow: Initial Lesion Type I: increase the numbers of macrophage and appearance of Foam Cells distributed at random Type II: (fatty streak) lesion: consist primarily of layers of macrophage foam cells and lipid-laden smooth muscle cells and include lesions grossly designated as fatty streaks Intermediary lesion (preatheroma) Type III: describe as a pathological intimal thickening with a poorly formed fibrous cap (because of the absence of a necrotic core). Typically, these lesions show incompletely coalesced extracellular lipid, most of which is located deep within the plaque, underneath a layer of macrophages and smooth muscle cells. Advance lesion Type IV: (atheroma) lesion: confluence of lipid collections creates an extracellular dense accumulation of fat in a well determined area of tunica intima in other word "a well-formed cellular cap overlying a confluent, necrotic, fatty core". Type V: (fibro-atheroma) lesion: thick layers of fibrous connective tissue (thick cellular caps) overlying a largely necrotic, fatty mass. Complicated lesion Type VI: complicated fibro-atheroma lesion: plaque with surface defect and/or hematoma/hemorrhage and/or thrombotic deposit 2.5 Measurement of samples 2.5.1 Measurement of serum total cholesterol(162) Reagents used are supplied by BIOLABO SA and the method is as following : Mix Reagent 1 ( buffer ) + Reagent 2 ( enzymes ), mix gently until complete dissolution ( approximately 2 min.). Reagent Blank Standard Assay 1 ml 1ml 1ml – – Demineralised water 0.1 ml Standard – 0.1 ml – Specimen – – 0.1 ml Wait for 10 min. and then record absorbencies at 500 nm against reagent blank. Calculate the result as follows : Result = 𝐴𝑏𝑠 (𝐴𝑠𝑠𝑎𝑦) 𝐴𝑏𝑠 (𝑠𝑡𝑎𝑛𝑑𝑎𝑟𝑑) × 200 ( Standard concentration) Composition of Reagents : R1 (Buffer) composed of the following: Phosphate buffer, Chloro-4-phenol, Sodium Cholate, Triton×100, and preservatives R2 ( Enzymes) composed of the following: Cholesterol oxydase (CO), Cholesterol esterase (CE), Peroxydase (POD), 4Amino-antipyrine (PAP) and PEG 6000) R3( standard) contains: Cholesterol 200mg./dl Principle(163): The principle is described by Allian C., the reaction scheme is as follow: Cholesterol esters → Cholesterol + O2 → 𝐶𝐸 𝐶𝑂 Cholesterol + free fatty acids Cholesten-4-one-3 + H2O2 2H2O2 + phenol + PAP → 𝑃𝑂𝐷 Quinoeimine (pink) + 4 H2O The absorbance of the colored complex (Quinoeimine), proportional to the amount of cholesterol in the specimen, is measured at 500nm. 2.5.2 Measurement of Triglycerides Concentration(164) Reagents used are supplied by BIOLABO SA and the method is as following : Mix Reagent 1 ( buffer ) + Reagent 2 ( enzymes ), mix gently until complete dissolution ( approximately 2 min.). Reagent Blank Standard Assay 1 ml 1ml 1ml – – Demineralised water 0.1 ml Standard – 0.1 ml – Specimen – – 0.1 ml Wait for 10 min. and then record absorbencies at 500 nm against reagent blank. Calculate the result as follows : Result = 𝐴𝑏𝑠 (𝐴𝑠𝑠𝑎𝑦) 𝐴𝑏𝑠 (𝑠𝑡𝑎𝑛𝑑𝑎𝑟𝑑) × 200 ( Standard concentration) Composition of Reagents: R1 (Buffer) composed of the following : Pipes, Mgnesium chloride, Chloro-4-phenol, and preservatives R2 ( Enzymes) composed of the following: Lipase, Peroxydase (POD), Glycerol-3-phosphate oxydase (GPO), Glycerol Kinase (GK), 4-Amino-antipyrine (PAP), and Adenosine triphosphate Na (ATP) R3( standard) contains: Glycerol equivalent to Triglycerides 200mg./dl Principle(165): The principle is described by Fossati and Prencipe. The Reaction scheme is as follow Triglycerides → 𝐿𝑖𝑝𝑎𝑠𝑒 Glycero+ ATP ⇔ 𝐺𝐾 glycerol + free fatty acids glycerol-3-phosphate + ADP 𝐺𝑃𝑂 Glycerol-3-phosphate + O2 ⇔ Dihydroxyacetone Phosphate + H2O2 𝑃𝑂𝐷 H2O2 + 4-Chlorophenol + PAP → Quinoeimine (pink) + H2O The absorbance of the colored complex (Quinoeimine), proportional to the amount of triglycerides in the specimen, is measured at 500nm. 2.5.3 Measurement of Serum HDL-C Concentration(166) The used reagents were supplied by Spinreact and the method as the following: Precipitation: 1-The following solutions are added into a centrifuge tube: R (µ L) 100 Sample (ml) 1.0 2- Solutions are mixed well, allowed to stand for 10 minutes at room temperature. 3- Centrifugation at 4000 rpm for 20 minute. 4- Then the supernatant is collected to test HDL-C. Test: HDL-C is measured by following the cholesterol reagent instructions. Calculations : With calibrator : 𝐴𝑏𝑠 (𝑆𝑎𝑚𝑝𝑙𝑒) 𝐴𝑏𝑠 (𝐶𝑎𝑙𝑖𝑏𝑟𝑎𝑡𝑜𝑟) × (Calibrator conc.) = mg/dl HDL-C in the sample. With factor : (Abs) sample x 320 = mg/dl HDL-C in the sample. Composition of Reagents : R Phosphotungstic acid 14 mmol/L Precipitating Reagent Magnesium chloride 2 mmol/L Optional Cholesterol Ref. 1001092 Ref. 1001093 Principle(166): The VLDL and LDL lipoproteins from serum or plasma are precipitated by phosphotungstate in the presence of magnesium ions. After removed by centrifugation the clear supernatant containing HDL is used for the determination of HDL cholesterol 2.5.4 Calculation of LDL, VLDL and Atherogenic index(77) LDL = Total cholesterol – (HDL + VLDL) VLDL = serum triglyceride / 5 Atherogenic index = (Total cholesterol – HDL-C)/HDL-C. 2. 5.5 Measurement of serum malondialdehyde (MDA) Level(167). The level of malonaldialdehyde was determined by modified procedure described by (Guidet B.and Shah S.V.,1989) Principle : The test is based on reaction of MDA with thiobarbituric acid (TBA); forming an MDA-TBA2product that absorb strongly at 532nm as follow Preparation of reagent: 1. Trichloroacetic acid reagent 70% 70gm of TCA was dissolved in a final volume of 100ml of DW 2. TCA reagent 17.5% 5ml of 70% was diluted upto 20ml with distall water 3. TBA reagent 0.6% 0.06gm of TBA was dissolved in final volume of 10ml of DW using a water bath for complete dissolving of TBA 1 Procedure : 1- 150ul of serum sample was poured in a test tube and 1ml of 17.5% TCA was added 2- 1ml of 0.06% TBA was added 3- The tube were mixed well by vortex ,incubated in boiling water bath for 15min, and then allow to cool 4- 1ml of 70% TCA was added 5- The mixture was left to stand at room temperature for 20min 6- The tube was centrifuge at 2000Xg for 15min ,and the supernatant was taken out for measuring spectrophotometriclly at 532nm Calculation of serum MDA: The concentration of MDA = 𝑎𝑏𝑠𝑜𝑟𝑏𝑎𝑛𝑐𝑒 ×𝐷 𝐿∗𝜀 L: Light bath (cm) 𝜀: Extinction coefficient =1.56 × 105 M-1 Cm-1 D: Dilution factor = 𝑣𝑜𝑙𝑢𝑚𝑒 𝑢𝑠𝑒𝑑 𝑖𝑛 𝑅𝑒𝑓.(𝑚𝑙) 0.15 = 1+1+1+0.15 0.15 = 21 2. 5.6 Determination of serum reduced glutathione (GSH) Principle(168): 5,5-dithiobis (2-nitrobenzoic acid) DTNB is a disulfide chromogen, that is, readily reduced by sulfahydryl group of GSH to intensely yellow compound . The absorbance of reduced chromogen is measured at 412nm directly proportional to the GSH concentration Preparation of reagents: 1- Precipitating solution (TCA 50%) 50gm of TCA were dissolved in final volume of 100ml of DW. 2- Ethylenediamine tetra-acetic acid-disodium (EDTA-Na2)(0.2M) 3.7224gm of EDTA-Na2 was dissolved in final volume of 50ml DW. 3- Tris –EDTA Buffer 0.4845gm of tris was dissolved in 8ml of DW. 1ml of 0.2 M EDTA-Na2 solutions was added and brought to final volume of 10ml with DW. The PH was adjusted to 8.9 by the addition of 1M of HCL .this solution is stable for at least 10days. 4- DTNB reagent (0.01M) 0.0396 g of DTNB was dissolved in absolute methanol, and brought to the final volume of 10ml .this solution is stable for 13 week at 4C 5-GSH standards solution (0.001M) Stock standards solution (0.001M) was prepared by dissolving 0.0156gm of GSH in final volume of 50ml of 0.2M EDTA solution, dilutions were made in EDTA solution to 5.10,25,50,100,200,300 &400uM Procedure: Duplicate of each standard and sample test tubes were prepared. Solution were mixed as in the following: Reagent Sample (uL) Reagent blank(uL) Standard (uL) Serum 100 - - Standard - - 100 DW 800 900 800 TCA 100 100 100 Tube were mixed in vortex mixer intermittently for 10-15min,and centrifuge for 15minat 3000xg,then pippetted into tubes. Reagent Sample (uL) Reagent blank(uL) Standard (uL) Supernatant 400 400 400 Tris-EDTA 800 800 800 DTNB reagent 20 20 20 Tubes were mixed in a vortex mixer. The spectrophotometer was adjusted with reagent blank to read zero absorbance (A) at 412nm and the absorbance of the standards and sample was read within 3min of the addition of the DYNB reagent Calculation of serum GSH: The concentration of serum GSH is obtained from the calibration curve in uM (figure ) abs 0.25 y = 0.0005x + 0.0089 R² = 0.9852 Absorbance 0.2 0.15 0.1 0.05 0 0 100 200 300 400 500 GSH Concentration uM 2.6 Measurement of aortic diameter, intima-media thickness and blood flow velocities of the aorta, renal artery and intra-renal arteries A triplex Doppler ultrasound machine (Siemens versa plus, made in Germany) which combines a B-mode ultrasound image with a pulse wave and color flow Doppler unit was used for examining rabbits. A 7.5 – 10 MHz linear array probe was used for this purpose. All rabbits were examine in private clinic by Dr. Akeel AM. H. Zwain PhD U.K. Lecturer in Cardiovascular Physiology. This machine was used for measuring aortic diameter and intima-media thickness and aortic, renal artery and intra-renal arteries blood flow velocities at the start of the study (at zero time). 4 rabbits were examined after 6 weeks for follow up ( i.e. when starting the treatment with sildenafil and vardenafil ), and again all rabbits were examined at the end of the study (after 12 weeks). The left lateral side of all rabbits was shaved by an electrical clipper. The rabbits were examined at morning after an overnight fasting to prevent the occurrence of intestinal gases which may prevent optimal visualizing of the arteries; the left lateral side was used for technical reasons . Rabbits were sedated by administration of 5mg/kg body weight diazepam intraperitoneally(169). Doppler angle was set at zero, the aorta was first visualized and identified, the diameter and intima- media thickness of the aorta ( at the origin of renal artery ) were measured. Then the aortic blood flow velocities ( peak systolic velocity and end diastolic velocity ) were measured electronically by the soft wave device built in the Doppler machine. The renal artery and intra renal arteries of the left kidney were visualized and identified using color flow mapping and their blood flow velocities ( peak systolic velocity and end diastolic velocity ) were also measured as mentioned earlier. 2.7 Calculation of Doppler indices(170) The Doppler indices calculated include the following 1-Calculation of resistive index ( RI): Resistive index (RI) was calculated from the following equation: RI = (𝑝𝑒𝑎𝑘 𝑠𝑦𝑠𝑡𝑜𝑙𝑖𝑐 𝑣𝑒𝑙𝑜𝑐𝑖𝑡𝑦(𝑃𝑆𝑉)−𝑒𝑛𝑑 𝑑𝑖𝑎𝑠𝑡𝑜𝑙𝑖𝑐 𝑣𝑒𝑙𝑜𝑐𝑖𝑡𝑦) 𝑝𝑒𝑎𝑘 𝑠𝑦𝑠𝑡𝑜𝑙𝑖𝑐 𝑣𝑒𝑙𝑜𝑐𝑖𝑡𝑦(𝑃𝑆𝑉) 2-Calculation of pulsatality index ( PI): Pulsatality index (PI) was calculated from the following equation: PI = (𝑝𝑒𝑎𝑘 𝑠𝑦𝑠𝑡𝑜𝑙𝑖𝑐 𝑣𝑒𝑙𝑜𝑐𝑖𝑡𝑦(𝑃𝑆𝑉)−𝑒𝑛𝑑 𝑑𝑖𝑎𝑠𝑡𝑜𝑙𝑖𝑐 𝑣𝑒𝑙𝑜𝑐𝑖𝑡𝑦 ) 𝑚𝑒𝑎𝑛 𝑣𝑒𝑙𝑜𝑐𝑖𝑡𝑦 The mean blood flow velocity (MV) is calculated from this equation(171) : MV = 1/3[peak systolic velocity +2(diastolic velocity)] 3-Calculation of renal-aortic ratio (RAR): This is the ratio of the peak systolic velocity (PSV) in the renal artery to the peak systolic velocity (PSV) in the aorta. So it is calculated by the following equation: RAR = 𝑃𝑆𝑉 𝑖𝑛 𝑟𝑒𝑛𝑎𝑙 𝑎𝑟𝑡𝑒𝑟𝑦 𝑃𝑆𝑉 𝑖𝑛 𝑎𝑜𝑟𝑡𝑎 2.8: Histopathological procedure The aortic arch with about 1 cm of the descending aorta was fixed in 10% formalin for 24 hour, then washed and dehydrated in ascending series of ethanol solution (70,80,90,95%) for 2hr. in each concentration and 4hr divided on two changes for 100% (v/v) , cleared in two changes of xylen, 15 minutes for each, then embedded in paraffin wax. Thin section about (6mm) was dewaxed in xylen for 6 minutes, hydrated in descending series of ethanol (2 min. for each 2 change in 100% ethanol, then 2 min. in each of 95, 90, 80 and 70% v/v of ethanol solution, then transferred to distilled water for 2 minutes. The sections were stained in haematoxylin for 2-5 min, washed in tap water for 2-3 min, discolored in 0.5-1% HCL in 70% alcohol for few seconds, washed in tap water for at least 5 min and stained in 1% aqueous eosin for 1-2 min., then the section washed in tap water and dehydrated in ascending series of ethanol ( 70,80,90,and 100% v/v ) for the same period of hydration, then cleared in xylen and mounted in DPX (172). 2.9: Statistical analysis Statistical analyses were performed using SPSS 12.0 for windows.lnc. Data were expressed as mean ± SEM.; paired t-test was used to compare the mean values within each group at different time. Analysis of Variance (ANOVA) was used for the multiple comparison among all groups followed by post-hoc tests using LSD method. Pearson correlation coefficient was used to assess the associations between two continuous variable of study parameter. Chi-square test was also used to compare histopathological changes in various groups. In all tests, P< 0.05 was considered to be statistically significant(173). 3.1 Effect on serum total cholesterol (TC) level In all groups of rabbits, the initial values of Serum TC were similar. For the first 6 weeks, serum levels of TC were significantly increased in groups (II, III, IV) fed with atherogenic diet(P<0.05) and remained unchanged in the Normal control group (I), as shown in table (3) and figure (1). After another 6 weeks on which groups (II, III, IV) continued to have cholesterol enriched diet, rabbits developed severe hypercholesterolemia with serum levels of TC further increased significantly (P<0.05). The changes in the levels of serum TC in the four groups are summarized in table (3) and figure(1). There was a statically significant difference(P<0.05) in serum levels of TC between the normal control group (I) and the groups on high cholesterol diet (II,III &IV). While The TC values showed insignificant difference between the induced untreated group (II),vardenafil treated group (III) & sildenafil treated group (IV), as shown in table (4). Table (3): Changes of serum TC level in mg/dl of the four experimental groups.(N = 6 rabbits in each group). The values are expressed as mean ±SEM. At zero time At 6 weeks At 12 weeks Normal control 48±2.1 50±2.6 49.4±2.3 Dietary induced untreated 47± 1.3 470±5.6* 710±9.4* Vardenafil 18mg./kg 46±1.7 510±4.5* 690±5.8* 47.5±2.2 495±3.5* 745±6.4* Sildenafil 5mg./kg * p<0.05 Table (4) : Multiple comparisons between different groups mean values of serum TC level in (mg/dl) using using ANOVA test Groups Vardenafil Sildenafil Induced untreated –640.6* –695.6* Induced untreated 20.0 -35.0 Sildenafil 55.0 Normal control * p<0.05 –660.6* normal dietary induced untreated vardenafil sildenafil 800 700 serum TC mg/dl 600 500 400 300 200 100 0 0 wk 6 wk 8 wk 10 wk 12 wk Figure (1): Effect of Vardenafil and Sildenafil on serum TC level in comparison to the two control groups (normal and induced untreated) 3.2 Effect on serum triglyceride (TG) level The basal levels of serum TG were similar in all groups of rabbits. But it increased significantly in groups (II, III, IV) fed with atherogenic diet(P<0.05) for the first 6 weeks, while unchanged in the normal control group (I), as shown in table (5) and figure (2). After 12 weeks, there was an increase in the serum TG level (P<0.05) in the induced untreated group, sildenafil treated group and vardenafil treated group which continued to have cholesterol enriched diet, as shown in table (5) and figure(2). Serum levels of TG showed significant difference(P<0.05) between the normal control group (I) and the groups on high cholesterol diet (II,III &IV). The TG values were not significantly different between the untreated high cholesterol diet (II) , vardenafil treated group(III) & sildenafil treated group (IV), as shown in table (6). Table (5): Changes of serum TG level in mg/dl of the four experimental groups.(N = 6 rabbits in each group). The values are expressed as mean ±SEM. At zero time At 6 weeks At 12 weeks Normal control 33±0.4 35±0.4 34±0.3 Dietary induced untreated 36±0.2 180±2.2* 210±3.4* Vardenafil 18mg./kg 38±0.6 175±2.4* 223±4.2* Sildenafil 5mg./kg 35±0.3 169±3.2* 205±2.6* * p<0.05 Table (6) : Multiple comparisons between different groups mean values of serum TG level in ( mg/dl) using ANOVA test Groups Vardenafil Sildenafil Induced untreated Normal control –189* –171* Induced untreated -13 5 Sildenafil -18 –176* * p<0.05 normal dietary induced untreated vardenafil sildenafil serum triglyceride in mg/dl 250 200 150 100 50 0 0 wk 6 wk 8 wk 10 wk 12 wk Figure(2): Effect of vardenafil 18mg/kg/day and sildenafil 5mg./kg/day treatment on serum TG level in comparison to the two control groups ( normal 3.3 Effect on HDL-C level Similar serum HDL-C level was found in the zero time. This level was significantly increased in groups (II, III, IV) fed with atherogenic diet for 6 weeks( P<0.05). Remarkably it remained unchanged in normal control group (I) as shown in table (7), and figure (3). At the end of treatment (12 weeks), serum HDL-C level in groups (II, III, IV) did not changed (P<0.05) as shown in table (7), and figure (3). It was found that serum levels of HDL-C between the normal control group (I) and the groups on high cholesterol diet (II,III &IV) significantly differ, while The HDL-C values were not significantly different between the untreated high cholesterol diet(II), vardenafil treated group (III) & sildenafil treated group (IV), as shown in table (8). Table (7): Changes of serum HDL-C level in mg/dl of the four experimental groups.(N = 6 rabbits in each group). The values are expressed as mean ±SEM. At zero time At 6 weeks At 12 weeks Normal control 16.2±0.3 16.6±0.2 16.3±0.3 Dietary induced untreated 15.8±0.2 17.8±0.3* 19.1±0.2* Vardenafil 18mg./kg 15.6±0.4 18.6±0.2* 18.5±0.4* Sildenafil 5mg./kg 16.5±0.5 18.5±0.4* 19.6±0.4* * p<0.05 Table (8) : Multiple comparisons between different groups mean values of serum HDL-C level in ( mg/dl) using ANOVA test Groups Vardenafil Sildenafil Induced untreated –2.2* –3.3* Induced untreated 0.6 -0.5 Sildenafil 1.1 Normal control * p<0.05 -2.8* normal dietary induced untreated vardenafil sildenafil 20 serum HDL-C in mg/dl 17.5 15 12.5 10 zero time 6 wk 8 wk 10 wk 12 wk Figure (3): Effect of Sildenafil 5mg/kg/day and Vardenafil 18mg./kg/day treatment on serum HDL-C level in comparison to the two control group (normal and induced untreated) 3.4 Effect on serum LDL-C level At the zero time, the values of serum LDL-C were the same in all groups of rabbits. Serum LDL-C level increased significantly in groups (II, III, IV) fed with atherogenic diet for 6 weeks (P<0.05) as illustrated in table (9) and figure (4). For the another 6 weeks, serum LDL-C level goes increase in groups (II, III, IV) fed with atherogenic diet(P<0.05) as shown in table (9) and figure (4). There was a statically significant difference (P<0.05) in serum levels of LDL-C between the normal control group (I) and the groups on high cholesterol diet (II,III &IV). While the LDL-C values did not change significantly among the untreated high cholesterol diet(II), vardenafil treated group (III) and sildenafil treated group (IV), as shown in table (10). Table (9): Changes of serum LDL-C level in mg/dl of the four experimental groups.(N = 6 rabbits in each group). The values are expressed as mean ±SEM. At zero time At 6 weeks At 12 weeks Normal control 25.2±1.7 26.6±2.25 26.5±1.5 Dietary induced untreated 23.3±2.26 418.0±4.5* 652.4±8.5* Vardenafil 18mg./kg 22.9±1.6 459.0±4.9* 629.0±2.8* Sildenafil 5mg./kg 23.5±1.9 444.7±4.5* 687.0±3.9* * p<0.05 Table (10) : Multiple comparisons between different groups mean values of serum LDL-C level in ( mg/dl) using ANOVA test Groups Vardenafil Sildenafil Induced untreated Normal control Induced untreated Sildenafil –602.5* –660.5* 23.4 -34 –625.9* 58 * p<0.05 800 700 serum LDL-C mg/dl 600 500 400 300 200 100 0 zero time Figure (4): Effect of 6 wk 8 wk 10 wk 12 wk vardenafil 18mg/kg/day and sildenafil 5mg./kg/day 3.5 Effect on VLDL-C level The initial values of serum VLDL-C were similar in all groups of rabbits. After 6 weeks, serum levels of VLDL-C were significantly increased in groups (II, III, IV) fed with atherogenic diet(P<0.05) and remained unchanged in the normal control group (I) as shown in table (11), and figure (5). After 12 weeks on the high-cholesterol diet, serum levels of VLDL-C further increased in groups (II, III, IV) (P<0.05). The changes in the levels of serum VLDL-C in the four groups are summarized in table (11) and figure (5). There was a statistically significant difference(P<0.05) in serum levels of VLDL-C between the normal control group (I) and the groups on high cholesterol diet (II,III &IV). While the VLDL-C values were not significantly different between the untreated high cholesterol diet(II) and vardenafil treated group (III) and sildenafil treated group (IV), as shown in table (12). Table (11): Changes of serum VLDL-C level in mg/dl of the four experimental group s.(N = 6 rabbits in each group). The values are expressed as mean ±SEM. At zero time At 6 weeks At 12 weeks Normal control 6.6±0.08 7.0±0.08 6.8±0.06 Dietary induced untreated 7.2±0.04 36.0±0.44* 42.0±0.68* Vardenafil 18mg./kg 7.6±0.12 35.0±0.48* 44.6±0.84* Sildenafil 5mg./kg 7.0±0.06 33.8±0.64* 41.0±0.52* * p<0.05 Table (12) : Multiple comparisons between different groups mean values of VLDL-C level using ANOVA test Groups Vardenafil Sildenafil Induced untreated –37.8* –34.2* Induced untreated -2.6 1.0 Sildenafil -3.6 Normal control * p<0.05 –35.2* normal dietary induced untreated vardenafil sildenafil 50 45 serumVLDL-C inmg/dl 40 35 30 25 20 15 10 5 0 zero time 6 wk 8 wk 10 wk 12 wk Figure (5): Effect of vardenafil 18mg/kg/day and sildenafil 5mg./kg/day treatment on serum VLDL-C level in comparison to the two control groups (normal and induced untreated) 3.6 Effect on atherogenic index Initially, the atherogenic index values were similar in all groups of rabbits. There was significant increase in atherogenic index (P<0.05) in groups (II, III, IV) fed with atherogenic diet for 6 weeks as shown in table (13), and figure (6). After another 6 weeks, a statistically significant increase in atherogenic index was noted in the induced untreated group, sildenafil treated group and vardenafil treated group continued to have atherogenic diet(P<0.05), as listed in table (13), and figure (6). The Atherogenic index value of normal control group (I) was statically significant differed (P<0.05) from the high cholesterol diet (II,III &IV). While The Atherogenic index values were not significantly different between the untreated high cholesterol diet(II), vardenafil treated group (III) and sildenafil treated group (IV), as shown in table (14). Table (13): Changes of atherogenic index in of the four experimental groups.(N = 6 rabbits in each group). The values are expressed as mean ±SEM. At zero time At 6 weeks At 12 weeks Normal control 1.96±0.15 2.05±0.22 2.07±0.12 Dietary induced untreated 1.85±0.18 28.65±0.49* 44.51±0.99* Vardenafil 18mg./kg 2.00±0.17 30.88±0.71* 41.07±0.23* Sildenafil 5mg./kg 1.79±0.16 29.00±1.51* 43.35±0.17* * p<0.05 Table (14) : Multiple comparisons between different groups mean values of atherogenic index using ANOVA test Groups Vardenafil Sildenafil Induced untreated –39.0* –41.28* Induced untreated 3.44 1.16 Sildenafil 2.28 Normal control -42.44* * p<0.05 normal dietary induced untreated vardenafil sildenafil 50 45 atherogenic index 40 35 30 25 20 15 10 5 0 zero time Figure (6): Effect of 6 wk 8 wk 10 wk 12 wk vardenafil 18mg/kg/day and sildenafil 5mg./kg/day 3.7 Effect on serum level of MDA Before the experiment, the baseline levels of serum MDA were not significantly different among all groups. After 6 weeks of high cholesterol diet, the MDA level significantly increased in induced untreated group (II) (P<0.05) as well as sildenafil treated group (III) & vardenafil treated group (IV) (P<0.05). After 12 weeks of high cholesterol diet, there was a further significant increase in induced untreated (II) (P<0.05), while sildenafil treated group (III) and vardenafil treated group (IV) were not significantly different at (P<0.05), but it remained greater than the normal values. The changes in MDA level are summarized in table (15) and figure (7). The serum MDA level of sildenafil treated (III) and vardenafil treated group (IV) was significantly (P<0.05) lower than that of induced untreated group (II). There was no significant difference in MDA level between sildenafil treated group (III) and vardenafil treated group (IV), as shown in table (16) Table (15): Sequential changes of serum MDA level mmole/L of the four experimental group. The data expressed as Mean ±SEM (N=6 in each group) Using paired T-test. At zero time At 6 weeks At 8 weeks At 10 weeks At 12 weeks Normal control 2.1±0.10 2.4±0.20 2.6±0.10 2.5±0.21 2.4±0.20 Dietary induced untreated 2.5±0.20 3.4±0.30 3.9±0.40 4.5±0.43* 5.9±0.45* Vardenafil 18mg./kg 2.6±0.30 3.5±0.41 3.7±0.35 3.9±0.30 4.1±0.22 Sildenafil 5mg./kg 2.4±0.25 3.3±.40 3.8±0.36 3.6±0.35 3.9±0.30 * p<0.05 Table (16): Multiple comparisons among different groups mean values of serum MDA level mg/dl using ANOVA TEST Groups Vardenafil Sildenafil Induced untreated Normal control –1.7 –1.5 Induced untreated 1.8* 2.0* Sildenafil -0.2 * p<0.05 -3.5* normal dietary induced untreated vardenafil sildenafil 6 serum MDA level mmol/l 5 4 3 2 1 0 zero time 6 wk 8 wk 10 wk 12 wk Figure (7) : the change in MDA Level in the four experimental group 3.8 Effect on serum reduced glutathione level GSH At zero time, the baseline levels of serum GSH were not significantly different among all groups. After 6 weeks of high cholesterol diet, the GSH level significantly decrease in induced untreated group (II) (P<0.05), sildenafil treaded group (III) (P<0.05) & vardenafil treated group (IV) (P<0.05). After 12 weeks of high cholesterol diet, there was a further significant decrease in induced untreated (II) (P<0.05), while sildenafil treat group (III) and vardenafil treated group (IV) not significantly different at (P<0.05). The changes in GSH level are summarized in table (17) and figure (8). The serum GSH level of sildenafil treat group (III) and vardenafil treated group (IV) was significantly (P<0.05) higher than that of induced untreated group (II). There was no significant difference in GSH level between sildenafil treat group (III)and vardenafil treated group (IV), as shown in table (18) Table (17): Sequential changes of serum GSH level uM of the four experimental groups. The data expressed as Mean ±SEM (N=6 in each group) Using paired T-test. At zero time At 6 weeks At 8 weeks At 10 weeks At 12 weeks Normal control 60±2.2 59±2.4 62±2.1 65±2.2 58±2.1 Dietary induced untreated 62±2.4 45±2.1 39±1.8 33±1.7* 29±1.6* Vardenafil 18mg./kg 58±2.0 42±2.0 39±1.9 37±1.8 40±1.8 Sildenafil 5mg./kg 63±2.5 44±2.1 40±2.0 38±2.0 39±1.7 * p<0.05 Table (18): Multiple comparisons among different groups mean values of serum GSH level uM using ANOVA TEST Groups Vardenafil Sildenafil Induced untreated Normal control Induced untreated Sildenafil * p<0.05 18 19 -11* -10* -1 29* normal dietary induced untreated vardenafil sildenafil 70 serum GSH level in mmol/l 60 50 40 30 20 10 0 zero time 6 wk 8 wk 10 wk 12 wk Figure (8): the change in Serum GSH Level in the four experimental groups 3.9 Effect on aortic intima-media thickness (IMT) At the end of 12 weeks of the study ,there was a statistically significant increase (P<0.05) in aortic intima-media thickness (IMT) in the induced untreated group, vardenafil treated group and sildenafil treated group while remained unchanged in the Normal control group (I), as shown in table (19) and figure (9). The increase in IMT in vardenafil and sildenafil treated groups was found to be lesser than that of induced untreated group, but there were no statistically significant differences (P<0.05) in the means of IMT of induced untreated group and treated groups (vardenafil and sidenafil groups), as shown in table (20). Table (19): Effect of atherogenic diet, Vardenafil 18mg/kg/day and Sildenafil 5mg/kg/day treatment on the rabbits aortic intima-media thickness measured in (mm) (No. = 6 rabbits in each group). At zero time At 12 weeks P-Value Normal control 0.32±0.01 0.32±0.03 P>0.05 Dietary induced untreated 0.31±0.02 0.58±0.02 P<0.05 Vardenafil 18m/kg 0.30±0.022 0.56±0.02 P<0.05 Sildenafil 5mg/kg 0.34±0.021 0.57±0.01 P<0.05 Table (20) : Multiple comparisons between different groups mean values of rabbits aortic intima-media thickness in measured in (mm) by using ANOVA test Groups Vardenafil Sildenafil Induced untreated -0.24* -0.25* - 0.26* Induced untreated 0.02 0.01 Sildenafil 0.01 Normal control *P<0.05 zero time 12 weeks aortic intima-media thickness 0.6 0.5 0.4 0.3 0.2 0.1 0 normal dietary induced untreated ardenafil sildenafil Figure (9): Effect of Vardenafil 18mg/kg/day and Sildenafil 5mg/kg/day treatment on rabbits aortic intima-media thickness (IMT) measured in (mm) in comparisons to the two control group (normal and induced untreated) 3.10 Effect on aortic diameter At the end of 12 weeks of the study ,there was a statistically significant increase (P<0.05) in aortic diameter in the induced untreated group, vardenafil treated group and sidenafil treated group and remained unchanged in the normal control group (I), as shown in table (21) and figure (10). The increase in aortic diameter in vardenafil and sidenafil treated groups was found to be close to that of induced untreated group, but there was no statistically significant differences (P<0.05) in the means of aortic diameter of induced untreated group and treated groups (vardenafil and sildenafil groups),as shown in table (22). Table (21): Effect of atherogenic diet, vardenafil 18mg/kg/day and sildenafil5mg/kg/day treatment on rabbits aortic diameter measured in (mm) (No. = 6 rabbits in each group). At zero time At 12 weeks P-Value Normal control 2.10 ± 0.08 2.28 ± 0.07 p>0.05 Dietary induced untreated 2.20± 0.17 2.70 ± 0.04 P<0.05 Vardenafil 18m/kg 2.17 ± 0.17 2.65 ± 0.07 P<0.05 Sildenafil 5mg/kg 2.11 ± 0.15 2.59 ± 0.05 P<0.05 Table (22): Multiple comparisons between different groups mean values of rabbits aortic diameter measured in (mm) by using ANOVA test Groups Vardenafil Sildenafil Induced untreated Normal control -0.37* -0.31* Induced untreated 0.05 0.11 Sildenafil -0.06 - 0.42* *P<0.05 zero time 12 weeks aortuc diameter in mm 3 2.5 2 1.5 1 0.5 0 normal dietary induced untreated vardenafil sildenafil Figure (10): Effect of vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on rabbits aortic diameter measured in (mm) in comparisons to the 3.11 Effect on aortic peak systolic blood flow velocity (PSV) At the end of 12 weeks of the study, there was an increase in aortic PSV in the induced untreated group and sidenafil treated group and there was a decrease in aortic PSV in vardenafil treated group and remained unchanged in the normal control group. But all these changes were statistically not significant (P>0.05), as shown in table(23) and figure (11). There was no statistically significant differences (P>0.05) in the means of aortic PSV of different groups as shown in table (24). Table (23): Effect of atherogenic diet, vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on the rabbits aortic peak systolic blood flow velocity measured in (cm/s) (No. = 6 rabbits in each group). At zero time At 12 weeks P-Value Normal control 29.0±2.1 31.1±2.3 p>0.05 Dietary induced untreated 30.0±2.4 32.0±3.0 p>0.05 Vardenafil 18m/kg 30.5±3.1 28.5±3.2 p>0.05 Sildenafil5mg/kg 30.2±2.9 31.4±2.6 p>0.05 Table (24): Multiple comparisons between different groups mean values of rabbits aortic peak systolic blood flow velocity measured in (cm/s) by using ANOVA test Groups Vardenafil Sildenafil Induced untreated Normal control 2.6 -0.3 Induced untreated 3.5 0.6 Sildenafil 2.9 - 0.9 aortic PSV in cm/sec zero time 12 week 39.5 38 36.5 35 33.5 32 30.5 29 27.5 26 24.5 23 21.5 20 normal dietary induced untreated vardenafil sildenafil Figure (11): Effect of vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on rabbits aortic peak systolic velocity (PSV) measured in (cm/s) in comparisons to the two control group ( normal and induced untreated) 3.12 Effect on aortic end diastolic blood flow velocity (EDV) At the end of 12 weeks of the study, there was a increase in aortic EDV in the induced untreated group and there was a decrease in aortic EDV in the vardenafil treated group, and sildenafil treated group. But all these changes were statistically not significant (P>0.05) as shown in table (25) and figure (12). There was no statistically significant difference (P>0.05) in the means of aortic EDV of different groups, as shown in table (26). Table (25): Effect of atherogenic diet, vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on the rabbits aortic end diastolic blood flow velocity measured in (cm/s) (No. = 6 rabbits in each group). At zero time At 12 weeks P-Value Normal control 4.2 ±0.56 4.7±0.62 p>0.05 Dietary induced untreated 4.6 ±1.2 5.2±0.36 p>0.05 Vardenafil 18m/kg 5.2 ±0.8 4.9±0.54 p>0.05 Sildenafil 5mg/kg 3.9 ±0.48 3.75±0.68 p>0.05 Table (26): Multiple comparisons between different groups mean values of rabbits aortic end diastolic blood flow velocity measured in (cm/s) by using ANOVA test Groups Vardenafil Sildenafil Induced untreated Normal control -0.2 0.95 -0.5 Induced untreated 0.3 1.45 Sildenafil -1.15 Zero time 12 week aortic EDV in cm/sec 6 5 4 3 2 1 0 Normal Dietary Induced Untreated Vardenafil Sildenafil 3.13 Effect on aortic blood flow resistive index (RI) At the end of 12 weeks of the study, there was a significant increase (P>0.05) in aortic blood flow RI in the induced untreated group, sildenafil treated group and vardenafil treated group, and remained unchanged in the normal control group, as shown in table (27) and figure (13). There was only statistically significant difference (P<0.05) between the means of aortic RI of normal control group and induced untreated group, sildenafil treated group and vardenafil treated group. No statistically significant differences (P>0.05) were observed between means of aortic RI of induced untreated group, sildenafil treated group and vardenafil treated group, as shown in table (28). Table (27): Effect of atherogenic diet, vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on the rabbits aortic blood flow resistive index (No. = 6 rabbits in each group). At zero time At 12 weeks P-Value Normal control 0.82±0.019 0.84±0.02 p>0.05 Dietary induced untreated 0.83±0.022 0.91±0.021 P<0.05 Vardenafil 18m/kg 0.83±0.034 0.9±0.021 P<0.05 Sildenafil5mg/kg 0.85±0.026 0.89±0.031 P<0.05 Table (28): Multiple comparisons between different groups mean values of rabbits aortic blood flow resistive index by using ANOVA test Groups Vardenafil Sildenafil Induced untreated Normal control -0.06* -0.05* Induced untreated 0.01 0.02 Sildenafil -0.01 *P<0.05 - 0.07* zero time 0.92 12 weeks Aortic blood flow RI 0.9 0.88 0.86 0.84 0.82 0.8 0.78 0.76 normal dietary induced untreated vardenafil sildenafil Figure (13): Effect of vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on rabbits aortic blood flow resistive index (RI) in comparisons to the two control group (normal and induced untreated) 3.14 Effect on aortic blood flow pulsatality index (PI) At the end of 12 weeks of the study, there was a significant increase (P>0.05) in aortic blood flow PI in the induced untreated group, sildenafil treated group and vardenafil treated group, and remained unchanged in the normal control group, as shown in table (29) and figure (14). There was only statistically significant difference (P<0.05) between the means of aortic PI of normal control group and induced untreated group, sildenafil treated group and vardenafil treated group. No statistically significant differences (P>0.05) were observed between means of aortic PI of induced untreated group, sildenafil treated group and vardenafil treated group, as shown in table (30). Table (29): Effect of atherogenic diet, vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on the rabbits aortic blood flow pulsatality index (No. = 6 rabbits in each group). At zero time At 12 weeks P-Value 1.95±0.1 1.83±0.11 p>0.05 Dietary induced untreated 1.88±0.097 2.9±0.079 P<0.05 Vardenafil 18m/kg 1.9±0.093 2.77±0.15 P<0.05 Sildenafil5mg/kg 2.01±0.16 2.85±0.11 P<0.05 Normal control Table (30): Multiple comparisons between different groups mean values of rabbits aortic blood flow pulsatality index by using ANOVA test Groups Vardenafil Sildenafil Induced untreated -0.94* -1.02* - 1.07* Induced untreated 0.31 0.05 Sildenafil 0.08 Normal control *P<0.05 zero time 12 weeks 3 Aortic flow PI 2.5 2 1.5 1 0.5 0 normal dietary induced untreated vardenafil sildenafil Figure (14): Effect of vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on rabbits aortic blood flow pulsatality index (PI) in comparisons to 3.15 Effect on renal artery peak systolic blood flow velocity (PSV) At the end of 12 weeks of the study, there was statistically significant increase (P<0.05) in renal artery PSV in the induced untreated group, but no statistically significant changes (P>0.05) were observed in other groups, as shown in table (31) and figure (15). There was statistically significant difference (P<0.05) between the means of renal artery PSV of the induced untreated group and other three groups, as shown in table (32). Table (31): Effect of atherogenic diet, vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on the rabbits renal artery peak systolic velocity measured in cm/s (No. = 6 rabbits in each group). At zero time At 12 weeks P-Value Normal control 45.2±4.6 46.1±5.6 p>0.05 Dietary induced untreated 46.1±3.4 69.0±8.5 P<0.05 Vardenafil 18m/kg 44.5±4.1 58.0±4.5 p>0.05 Sildenafil 5mg/kg 46.0±4.2 57.0±3.5 p>0.05 Table (32): Multiple comparisons between different groups mean values of rabbits renal artery peak systolic blood flow velocity measured in cm/s by using ANOVA test . Groups Vardenafil Sildenafil Induced untreated Normal control -11.9 -10.9 Induced untreated 11* 12* Sildenafil *P<0.05 -1 - 22.9* zero time 12weeks 70 renal artery PSV cm/sec 60 50 40 30 20 10 0 normal dietary induced untreated vardenafil sildenafil Figure (15): Effect of vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on rabbits renal artery peak systolic velocity (PSV) measured in cm/s in comparisons to the two control group (normal and induced untreated) 3.16 Effect on renal artery end diastolic blood flow velocity (EDV) At the end of 12 weeks of the study, there was a statistically significant decrease (P<0.05) in renal artery EDV in the vardenafil and sildenafil treated groups, but no statistically significant changes (P>0.05) were observed in normal group and induced untreated group, as in table (33) and figure (16). There was no statistically significant difference (P<0.05) between the means of renal artery EDV of the vardenafil treated group and sildenafil treated group, as shown in table (34). Table (33): Effect of atherogenic diet, , vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on the rabbits renal artery end diastolic velocity measured in cm/s (No. = 6 rabbits in each group). At zero time At 12 weeks P-Value Normal control 23.0±2.4 24.1±2.8 p>0.05 Dietary induced untreated 22.6±2.3 23.3±4.0 p>0.05 Vardenafil 18m/kg 23.9±2.1 18.2±1.8 P<0.05 Sildenafil5mg/kg 24.6±1.8 19.0±1.2 P<0.05 Table (34): Multiple comparisons between different groups mean values of rabbits renal artery end diastolic blood flow velocity measured in cm/s by using ANOVA test Groups Vardenafil Sildenafil Induced untreated Normal control 5.9* 5.1* Induced untreated 5.1* 4.3* Sildenafil 0.8 0.8 *P<0.05 zerc time 12weeks renal artery EDV in cm/sec 25 20 15 10 5 0 normal dietary induced untreated vardenafil sildenafil Figure (16): Effect of vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on rabbits renal artery end diastolic velocity (EDV) measured in cm/s 3.17 Effect on renal artery blood flow resistive index (RI) At the end of 12 weeks of the study ,there was a statistically significant increase (P<0.05) in renal artery blood flow RI only in the induced untreated group. No statistically significant changes (P>0.05) were observed in other groups, as shown in table (35) and figure (17). There was a statistically significant difference (P<0.05) between the means of renal artery RI of the induced untreated group and other three groups, as shown in table (36). Table (35): Effect of atherogenic diet, , vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on the rabbits renal artery blood flow resistive index (No. = 6 rabbits in each group). At zero time At 12 weeks P-Value Normal control 0.478 ± 0.024 0.480 ± 0.01 p>0.05 Dietary induced untreated 0.461± 0.013 0.647 ± 0.043 P<0.05 Vardenafil 18m/kg 0.482 ± 0.021 0.522 ± 0.025 p>0.05 Sildenafil 5mg/kg 0.496 ± 0.022 0.539 ± 0.027 p>0.05 Table (36): Multiple comparisons between different groups mean values of rabbits renal artery blood flow resistive index by using ANOVA test Groups Vardenafil Sildenafil Induced untreated Normal control -0.042 -0.059 - 0.167* Induced untreated 0.125* 0.108* Sildenafil 0.017 *P<0.05 zero time 12 weeks 0.7 Renal artery RI 0.6 0.5 0.4 0.3 0.2 0.1 0 normal dietary induced untreated vardenafil sildenafil Figure (17): Effect of vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on rabbits renal artery blood flow resistive index (RI) in comparisons to the two control group (normal and induced untreated) 3.18 Effect on renal pulsatality index (PI) artery blood flow At the end of 12 weeks of the study, there was statistically significant increase (P<0.05) in renal artery blood flow PI only in the induced untreated group. No statistically significant changes (P>0.05) were observed in other groups, as shown in table (37) and figure (18). There was statistically significant difference (P<0.05) between the means of renal artery PI of induced untreated group and other three groups, as shown in table (38). Table (37): Effect of atherogenic diet, , vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on the rabbits renal artery blood flow pulsatality index (No. = 6 rabbits in each group). At zero time At 12 weeks P-Value Normal control 0.74±0.057 0.72±0.021 p>0.05 Dietary induced untreated 0.68±0.026 1.12±0.13 P<0.05 Vardenafil 18m/kg 0.79±0.055 0.837±0.06 p>0.05 Sildenafil 5mg/kg 0.76±0.054 0.85±0.059 p>0.05 Table (38): Multiple comparisons between different groups mean values of rabbits renal artery blood flow pulsatality index by using ANOVA test Groups Vardenafil Sildenafil Induced untreated Normal control -0.117 -0.13 - 0.4* Induced untreated 0.283* 0.27* Sildenafil -0.013 *P<0.05 zero time 12 weeks 1.2 Renal artery PI 1 0.8 0.6 0.4 0.2 0 normal dietary induced untreated vardenafil sildenafil 3.19 Effect on intra-renal arteries peak systolic blood flow velocity (PSV) At the end of 12 weeks of the study, there was statistically significant increase (P<0.05) in intra-renal arteries PSV in the induced untreated group, but no statistically significant changes (P>0.05) were observed in other groups, as shown in table (39) and figure (19). There was statistically significant difference (P<0.05) between the means of renal artery PSV of the induced untreated group and other three groups, as shown in table (40). Table (39): Effect of atherogenic diet, , vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on the rabbits intra- renal arteries peak systolic velocity measured in cm/s (No. = 6 rabbits in each group). At zero time At 12 weeks P-Value Normal control 41.6±6.7 42.4±6.6 p>0.05 Dietary induced untreated 41.2±±3.4 64.1±7.8 P<0.05 Vardenafil 18m/kg 42.2±5.5 48.3±4.2 p>0.05 Sildenafil 5mg/kg 40.7±2.7 49.1±2.3 p>0.05 Table (40): Multiple comparisons between different groups mean values of the rabbits intra- renal arteries peak systolic velocity measured in cm/s by using covariance ANOVA test Groups Vardenafil Sildenafil Induced untreated Normal control Induced untreated Sildenafil *P<0.05 -5.9 -6.7 15.8* 15* 0.8 - 21.7* Intra-renal artery PSV in cm/sec zero time 12 weeks 70 60 50 40 30 20 10 0 normal dirtary induced untreated vardenafil sildenafil Figure (19): Effect of vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on the rabbits intra- renal arteries peak systolic velocity (PSV) measured in cm/s in comparisons to the two control group (normal and induced untreated) 3.20 Effect on intra-renal arteries end diastolic blood flow velocity (EDV) At the end of 12 weeks of the study, there was statistically significant decrease (P<0.05) in intra-renal arteries EDV in the vardenafil and sildenafil treated groups, but no statistically significant changes (P>0.05) were observed in normal group and induced untreated group, as shown in table (41) and figure (20). There was no statistically significant difference (P<0.05) between the means of intra-renal arteries EDV of the vardenafil treated group and sildenafil treated group, as shown in table (42). Table (41): Effect of atherogenic diet, vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on the rabbits intra- renal arteries end diastolic velocity measured in cm/s (No. = 6 rabbits in each group). At zero time At 12 weeks P-Value Normal control 22 ± 0.62 23.1 ±0.51 p>0.05 Dietary induced untreated 22.3 ±0.36 24.2 ±0.43 p>0.05 Vardenafil 18m/kg 22.4 ±0.41 19 ±0.32 P<0.05 Sildenafil 5mg/kg 21.9 ±0.32 18.4 ±0.37 P<0.05 Table (42): Multiple comparisons between different groups mean values of rabbits intra- renal arteries end diastolic velocity measured in cm/s by using ANOVA test Groups Vardenafil Sildenafil Induced untreated Normal control 4.1* 4.7* -1.1 Induced untreated 5.2* 5.8* Sildenafil -0.6 *P<0.05 intra-renal artery EDV in cm/sec zero time 12weeks 25 20 15 10 5 0 normal dietary induced untreated vardenafil sildenafil 3.21 Effect on intra-renal arteries blood flow resistive index (RI) At the end of 12 weeks of the study ,there was statistically significant increase (P<0.05) in intra-renal arteries blood flow RI only in the induced untreated group. No statistically significant changes (P>0.05) were observed in other groups, as shown in table (43) and figure (21). There was statistically significant difference (P<0.05) between the means of intra-renal arteries RI of induced untreated group and other three groups, as shown in table (44). Table (43): Effect of atherogenic diet, vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on the rabbits intra-renal arteries blood flow resistive index (No. = 6 rabbits in each group). At zero time At 12 weeks P-Value Normal control 0.461 ± 0.022 0.462 ±0.018 p>0.05 Dietary induced untreated 0.472±0.016 0.63±0.031 P<0.05 Vardenafil 18m/kg 0.46±0.024 0.51 ±0.038 p>0.05 Sildenafil 5mg/kg 0.48 ±0.029 0.53 ±0.021 p>0.05 Table (44): Multiple comparisons between different groups mean values of rabbits intra-renal arteries blood flow resistive index by using ANOVA test Groups Vardenafil Sildenafil Induced untreated Normal control -0.048 -0.068 Induced untreated 0.12* 0.1* Sildenafil -0.02 *P<0.05 - 0.168* zero time 12 weeks 0.7 Intr-renal artery RI 0.6 0.5 0.4 0.3 0.2 0.1 0 normal dietary induced untreated vardenafil sildenafil Figure (21): Effect of vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on rabbits intra-renal arteries blood flow resistive index (RI) in comparisons to the two control group (normal and induced untreated) 3.22 Effect on intra-renal arteries blood flow pulsatality index (PI) At the end of 12 weeks of the study, there was statistically significant increase (P<0.05) in intra-renal arteries blood flow PI only in the induced untreated group. No statistically significant changes (P>0.05) were observed in other groups, as shown in table (45) and figure (22). There was statistically significant difference (P<0.05) between the means of intra-renal arteries PI of induced untreated group and other three groups, as shown in table (46). Table (45): Effect of atherogenic diet, , vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on the rabbits intra-renal arteries blood flow pulsatality index (No. = 6 rabbits in each group). At zero time At 12 weeks P-Value Normal control 0.75± 0.051 0.73±0.043 P>0.05 Dietary induced untreated 0.71±0.035 1.1±0.084 P<0.05 Vardenafil 18m/kg 0.78±0.055 0.83±0.083 P>0.05 Sildenafil 5mg/kg 0.77±0.063 0.86±0.044 P>0.05 Table (46): Multiple comparisons between different groups mean values of rabbits intra-renal arteries blood flow pulsatality index by using ANOVA test Groups Vardenafil Sildenafil Induced untreated Normal control -0. 1 -0.13 - 0.37* Induced untreated 0.27* 0.24* Sildenafil 0.03 *P<0.05 zero time 1.2 12 weeks Intra-renal artery PI 1 0.8 0.6 0.4 0.2 0 normal dietary induced untreated Figure (22): Effect of vardenafil sildenafil vardenafil 18mg/kg/day and sildenafil 5mg/kg/day 3.23 Effect on renal artery-to-aortic systolic velocity ratio (RAR) peak At the end of 12 weeks of the study ,there was statistically significant increase (P<0.05) in (RAR) in the induced untreated group, vardenafil treated group and sidenafil treated group and remained unchanged in the normal control group (I), as shown in table (47) and figure (23). The increase of (RAR) in vardenafil and sidenafil treated groups was found to be lesser than that of induced untreated group, but there were no statistically significant differences (P<0.05) in the means of (RAR) of induced untreated group and treated groups (vardenafil and sildenafil groups), as shown in table (48). Table (47): Effect of atherogenic diet, , vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on rabbits renal artery-to-aortic peak systolic velocity ratio (No. = 6 rabbits in each group). At zero time At 12 weeks P-Value Normal control 2.00 ± 0.24 2.19 ±0.25 p>0.05 Dietary induced untreated 1.99 ±0.23 2.78 ±0.17 P<0.05 Vardenafil 18m/kg 2.01 ±0.27 2.65 ±0.23 P<0.05 Sildenafil 5mg/kg 1.98 ±0.24 2.72 ±0.36 P<0.05 Table (48): Multiple comparisons between different groups mean values of rabbits renal artery-to-aortic peak systolic velocity ratio by using ANOVA test Groups Vardenafil Sildenafil Induced untreated Normal control -0. 46* -0.53* Induced untreated 0.13 0.06 Sildenafil 0.07 *P<0.05 - 0.59* zero time 12 weeks Renal artery/Aortic ratio 3 2.5 2 1.5 1 0.5 0 normal dietary induced untreated vardenafil sildenafil Figure (23): Effect of vardenafil 18mg/kg/day and sildenafil 5mg/kg/day treatment on rabbits renal artery-to-aortic peak systolic velocity ratio in comparisons to the two control group (normal and induced untreated) 3.24 Histopathological findings At the end of the study a cross section of normal rabbits aorta shows the normal appearance of all three arterial wall layers (intima, media and adventitia). The intima-media lining shows no thickening in all rabbits of this group (100%). There was statistically significant difference between normal control group and other three groups (P<0.05), as shown in table (49) & (50) and figure (24) & (25). While High cholesterol diet untreated had virtually a 100% involvement of aorta with different severity of atherosclerosis (all animals had a degree of atherosclerosis). 16.7% of the group had initial atherosclerotic lesion as a phase II, 33.33% of the group had intermediate atherosclerotic lesion as phase III and 50% of the group had advance atherosclerotic lesion (33.33%as phase IV, and 16.7% as phase V) . There was statistically significant difference between induced untreated group and normal control group (P<0.05), as shown in table (49) & (50) and figure (24) & (25). Together sildenafil treated group results in non significant reduction in severity of atherosclerotic lesion compared with induced untreated group. 16.7 % of rabbits treated with sildenafil had initial atherosclerotic lesion as a phase II, 50% of the group had intermediate atherosclerotic lesion appear as phase III, and 33.33% of the group had advance atherosclerotic lesion as phase IV. There was no complicated lesion in sildenafil treated group, as shown in table (49) & (50) and figure (24) & (25).In concert vardenafil treated group results in non significant reduction in severity of atherosclerotic lesion compared with induced untreated group. 33.33 % of rabbits treated with vardenafil had initial atherosclerotic lesion as a phase II, 33.33% of the group had intermediate atherosclerotic lesion appear as phase III, and 33.33% of the group had advance atherosclerotic lesion as phase IV. There was no complicated lesion in vardenafil treated group, as shown in table (49) & (50) and figure (24) & (25). Table (49): Demonstration of different aortic atherosclerotic phases for different rabbit groups at the end of 12 weeks of the study Phases of atherosclerotic lesion groups Normal Phase 1 Phase 2 Phase 3 NO. 6 0 0 0 % 100 0 0 NO. 0 0 % 0 Sildenafil NO. 5mg/kg/day Vardenafil Normal control NO.= 6 Dietary induced untreated Phase 4 Phase 5 Phase 6 0 0 0 0 0 0 0 1 2 2 1 0 0 16.7 33.33 33.33 16.7 0 0 0 1 3 0 0 % 0 0 16.7 50 33.33 0 0 NO. 0 0 2 2 2 0 0 % 0 0 33.33 33.33 33.33 0 0 NO.= 6 18mg/kg/day 2 120 standard diet high cholest diet sildenafil vardenafil Percentage of involvment 100 80 60 40 20 0 normal phase 1 phase 2 phase 3 phase 4 phase 5 Figure (24): Percentages of aortic involvement with different atherosclerotic phases. Table (50): Demonstration of different aortic atherosclerotic lesions for different rabbit groups at the end of 12 weeks of the study Normal Normal control NO= 6 NO. Initial lesion Intermediate lesion Advance lesion Complicated lesion 6 0 0 0 0 % 100 0 0 0 0 Dietary induced untreated NO= 6 NO. 0 1 2 2 1 % 0 16.7 33.33 33.33 16.7 Sildenafil NO. 0 1 3 2 0 5mg/kg/day % 0 16.7 50 33.33 0 Vardenafil NO. 0 2 2 0 18mg/kg/day % 0 33.33 33.33 0 2 33.33 120 stand diet high cholest diet sildenafil vardenafil Percentage of involvment 100 80 60 40 20 0 Normal Initia Intermedia Advance Complicat Figure (25): Percentages of aortic involvement with different atherosclerotic lesions. Lumen Adventitia Figure (26): A cross section of normal rabbit aorta. The section stained with haematoxylin and eosin (×10) Foam cells Lumen Figure(27): A cross section of hypercholesterolemic rabbit's aorta. The section stained with haematoxylin and eosin ( ×10) Layer of foam cells Figure (28) A cross section of hypercholesterolemic rabbit's aorta. The section stained with haematoxylin and eosin ( ×10) Intimal thickness Figure (29):A cross section from aorta shows a pathological intimal thickening. The section stained with haematoxylin and eosin ( ×10) Preatheroma lesion Absence of fatty core Figure (30):A cross section from aorta shows a pathological intimal thickening. The section stained with haematoxylin and eosin ( ×10) Foam cell Intimal thickness Figure (31): A cross section of hypercholesterolemic rabbit's aorta. The section stained with haematoxylin and eosin ( ×40) Thin layer of fibrous cap Necrotic fatty core Figure (32): A cross section from aorta shows a fibro-atheromatous plaque. The section stained with haematoxylin and eosin ( ×10) Figure (33): A B-mode ultrasound image showing a rabbit abdominal aorta with normal aortic wall thickness , D1 is the intima-media thickness = 0.3 mm , D2 is the aortic diameter = 2.1 mm Figure (34):A B-mode ultrasound image showing a rabbit abdominal aorta with an increased aortic thickness (sclerotic wall), but there is no apparent plaques , D1 is the intima-media thickness = 0.6 mm , D2 is the aortic diameter = 2.8 mm Plaque Figure (35): A B-mode ultrasound image showing a rabbit abdominal aorta with an increased aortic thickness (sclerotic wall), there also dissemination of plaques inside the aorta , D1 is the intima media thickness = 0.7 mm Figure (36): A Doppler wave form spectrum showing a rabbits aortic blood flow velocity, PSV=0.49 m/s , EDV=0.06 m/s RI=0.88 , S/D ratio=8.2 Figure (37): A Doppler wave form spectrum showing a rabbits renal artery blood flow velocity, PSV=0.48 m/s , EDV=0.17m/s, RI=0.58 , S/D ratio=2.4 Figure (38): A Doppler wave form spectrum showing a rabbits intra-renal artery blood flow velocity, PSV=0.49 m/s , EDV=0.20m/s, RI=0.59 , S/D ratio=2.5 4.1 Effect on Serum Lipid Profile 4.1.1 Effect of cholesterol enriched diet on serum lipid profile and atherogenic index The cholesterol-fed rabbit is a model frequently used for studying the development and modifications in the natural history of atherosclerosis(174). In the current study, feeding of atherogenic diet to rabbits for 12 weeks resulted in marked hypercholesterolemia in which serum TC, TG, LDL-C, HDL-C, VLDL and atherogenic index levels were found to be increased in agreement with changes in these parameters which have been reported earlier Romero et al. (2000)(175) ; Prasad et al, ( 2007)(176) and Nigris et al. (2008)(177) and these results might be attributed to exogenous cholesterol (atherogenic diet). 4.1.2 Effect of Sildenafil and Vardenafil treatment on serum lipid profile and atherogenic index In this study, there was insignificant changes in serum TC, TG, HDL, LDL, VLDL and atherogenic index levels (p<0.05) in sildenafil and vardenafil treated rabbits as compared with that in the induced untreated group. This may be due to that the change in lipid parameter induced by high cholesterol diet (2% cholesterol for 12 weeks) overrides any changes from sildenafil and vardenafil treatment. However Kimura et al. (2003) found no significant differences in lipid parameter in smokers treated with sildenafil(178). 4.2 Effect on oxidative stress 4.2.1 Effect of high cholesterol diet on oxidative stress In the present study, hypercholesterolemic atherosclerosis was associated with increases in the serum levels of the lipid peroxidation product MDA, and decrease in the level of GSH suggesting an increase in the levels or activity of oxygen radicals. MDA and GSH have been considered as specific indicators of oxidative status (160). Increases in serum MDA and a decrease in GSH in hypercholesterolemic rabbit were also observed by Jale et al. (2004)(179). Moreover, increases in the levels of serum MDA in high cholesterolfed rabbits have been reported previously (180,181). Hypercholesterolemia could increase levels of reactive oxygen species in various ways. Hypercholesterolemia increases cholesterol content of platelets, polymorphonuclear leukocytes (PMNs) and endothelial cells. This result would lead to increase in activated complements 3 and 5 (C3a and C5a), platelet activating factor, interleukin-1, and tumor necrosis factor, and synthesis of prostaglandins and leukotriene (182,183,184). Thus our study further confirms that the increase in oxidative stress and subsequent lipid peroxidation together with the decrease in the level of antioxidant may provide mechanistic answer for atherosclerosis induced by high fat diet in rabbit. 4.2.2 Effect of Sildenafil and Vardenafil on MDA and GSH level Sildenafil and vardenafil had significant effect on serum MDA and GSH levels. Hence it inhibited the increase of serum MDA induced in high cholesterol-fed rabbits suggesting decrease in ROS and subsequent lipid peroxidation. Also these drugs prevent GSH depletion in hypercholesterolemia rabbit, and thus, maintain antioxidant reserve which is crucial for vascular protection against lipid peroxidation. This finding is consistent with Abdollahi et al. (2003)(185) who found that the use of cAMP and cGMP phosodiesterase inhibitors, theophylline and sildenafil, prevented lead induced increased lipid peroxidation and also protected from decreased thiol groups content and total antioxidant power of the gland and secretions. The same trend of effects was observed in gland and saliva. The antioxidant activity of PDEIs can be explained by that first. It is increase total antioxidant capacity by increasing the activities of CAT, GSH-Px, and total SOD(186). Second, sildenafil increase nitric oxide which activates guanylate cyclase resulting in an increase of the conversion of GTP to cGMP and cGMP which mediates an anti-oxidant action(185). Third, sildenafil could reduce NAD(P)H oxidase expression and superoxide formation in corpus cavernosum of hypercholesterolemic rabbits (186). 4.3 Effect on aortic intima-media thickness 4.3.1 Effect of cholesterol enriched diet on aortic intima-media thickness A significant increase in aortic intima-media thickness (P<0.05) was found in rabbits fed with cholesterol enriched diet as compared with that of the normal control group. This result is in agreement with that reported by Michael et al. (1999)(187) and Roberto et al. (2004)(188) who examined rabbits by high-resolution magnetic resonance imaging (MRI) and found that the vessel wall thickness increased significantly in cholesterol fed rabbits. the increment in aortic intima-media thickness might be due to lipid deposition on aortic wall. 4.3.2 Effect of Sildenafil and Vardenafil treatment on aortic intimamedia thickness There was insignificant reduction of aortic intima-media thickness in sildenafil and vardenafil treated rabbits as compared with that in the induced untreated group. Although sildenafil and vardenafil have the ability to improve endothelial function, and possess antioxidant action. Additionally sildenafil reduces endothelial proliferation as mentioned by, (Heller et al.) who concluded that the antiproliferative effect of nitric oxide was independent of cGMP, which would exclude a cGMP-dependent mechanism involving the protein kinase G(189). This finding may be due to the fact that rabbits were continued to be fed with cholesterol enriched diet together with the treatment with sildenafil and verdenafil which may prevent the maximal effect of the drugs or due to low doses of drugs. Another reason for the non significant reduction in aortic intima-media thickness may be due to small sample size studied and short duration of treatment. 4.4 Effect on aortic diameter 4.4.1 Effect of cholesterol enriched diet on aortic diameter A significant increase of aortic diameter (P<0.05) was found in rabbits fed with cholesterol enriched diet as compared to that of the normal control group. our finding is consistent with Mohammad (2008)(77) who had found significant increase in aortic diameter (P<0.05) in rabbits fed with cholesterol enriched diet as compared to that of the normal control group. The increase in the aortic diameter in rabbits fed high cholesterol diet can be explained by the expansive (enlargement) arterial remodeling which was recognized as an important determinant in vascular pathology in which narrowing of the lumen is the predominant feature(190). It is known that blood vessels accommodate to the increase in blood flow by arterial growth (Hemodynamic stimuli like flow and circumferential stress induce arterial remodeling to achieve homeostasis of shear stress and wall tension, respectively)(191,192). However, a decade ago it was described in macaque monkeys that radial enlargement of vessels can also occur in response to progressive plaque growth(193). However, Glagov found that the lumen area of atherosclerotic human vessels remained constant until the percent stenosis exceeded 40%. At this point lumen diameter decreased, resulting in a restriction in flow. On the other hand he have surprising finding that atherosclerotic arterial lumen narrowing is not simply the result of enlargement of atherosclerotic lesions. He found instead that arteries remodel over a large range of changes in wall mass, increasing the external diameter in a manner that allows preservation of the arterial flow (glagoven)(194). 4.4.2 Effect of Sildenafil and Vardenafil treatment on aortic diameter In sildenafil and vardenafil treated rabbits, there was no significant change (P<0.05) in the aortic diameter in comparison to that of the induced untreated group(P>0.05). Data of (Pache et al., 2002) in a study without a control group also showed a 5.8% increase in venous and arterial diameter after administration of 50 mg of sildenafil. The peak of the changes, however, was observed at 30 min and by 120 min. Both arterial and venous diameters had returned to baseline values. Polak et al. (2003) did not observe any statistically significant effect on the retinal arterial diameters, and no effect of sildenafil was found on a flicker-induced vasodilatation in retinal arteries or veins. Furthermore, previous study in normal male volunteers Grunwald et al. (2002) did not reveal any statistically significant effect in retinal venous and arterial diameters after administration of sildenafil(195). This result may be due to that the increase in the aortic diameter in rabbits fed high cholesterol diet (which can be explained by the expansive arterial remodeling) will override the changes expected from sildenafil and vardenafil treatment. However hemodynamic studies suggest that sildenafil is a modest vasodilator. So it does not largely affect aortic diameter . 4.5 Effect on peak systolic blood flow velocity 4.5.1 Effect of cholesterol enriched diet on peak systolic blood flow velocity There was non-significant increase in aortic peak systolic velocity (P>0.05) but significant increase in renal artery and intra-renal arteries peak systolic velocity (P<0.05) in rabbits fed with cholesterol enriched diet as compared to that of the normal control group. This result is in agreement with that reported by Atsushi et al. (2003)(196) who found that the peak systolic velocity of atherosclerotic iliac arteries of rabbits increased significantly (2.1 times) in comparison to normal iliac arteries. The increase in renal artery and intra-renal arteries peak systolic velocity is due to the increased wall thickness and the development of plaques in the arterial wall by cholesterol enriched diet feeding. When plaques protrude in to the blood vessels, narrowing of vascular lumen occurs which leads to very high blood flow velocity through the stenosed region(147,148). Moreover, the increase in blood flow PSV may be due to the fact that the atherosclerotic vessels are hardened and exhibit stiff and non compliant state(197). The finding that the aortic PSV was not increased significantly can be explained by the fact that the aortic lumen is larger and it is difficult for plaques to cause stenosis in such artery with a wide lumen to affect blood flow velocity. Another observation is that the aortic PSV in some rabbits was found to decrease. This may be due to a possible development of heart failure which perhaps caused reduction of blood pumping capability of the heart and consequently reduction of aortic peak systolic velocity(146). 4.5.2 Effect of Sildenafil and Vardenafil treatment on peak systolic blood flow velocity In sildenafil and vardenafil treated rabbits there was a significant decrease (P<0.05) in the renal artery and intra-renal artery PSV in comparison to that of the induced untreated group, On the contrary, there was no significant change in the aortic PSV (P>0.05) found in sildenafil treated and vardenafil treated rabbits in comparison to the induced untreated group. These findings are in agreement with those reported by Ercan et al. (2005) who concluded that Sildenafil citrate had no significant effect on aortic and SMA circulation and only caused mild changes in the carotid artery circulation. These alterations may be considered clinically insignificant (198). Additionally this result is consistent with that observed by Ardicoglu et al. (2005) who demonstrated that peroral sildenafil citrate usage had slight effects on hemodynamic parameters of the lower segmental branch of right renal artery(199). The reduction of renal artery and intra-renal artery peak systolic velocities may be partly due to the fact that the treatment with The PDE5 inhibitor sildenafil and vardenafil has a double vascular muscle relaxant potential, that is an indirect mechanism dependent on NO-cGMP and a direct potential possibly via the inhibition of Ca2_ influx through receptor operated and voltage dependent Ca2_ channels(200). It is possible that the vasodilator action of sildenafil could potentially release endogenous mediators of reconditioning from endothelial cells, such as adenosine or bradykinin, which may trigger a signaling cascade through kinase action, resulting in NO synthase phosphorylation and NO release(200) . This could mean that sildenafil produces a positive effect by acting directly on the renal dysfunctional endothelium and improving its function but without collateral systemic effects (200). 4.6 Effect on End diastolic blood flow velocity 4.6.1 Effect of cholesterol enriched diet on end diastolic blood flow velocity There was unsignificant (P>0.05) decrease in aortic end diastolic velocity and non significant (P<0.05) increase in renal artery and intrarenal arteries end diastolic velocity in rabbits fed with cholesterol enriched diet as compared to that of the normal control group. This finding is consistent with Mohammad (2008)(77) who found that cholesterol enriched diet results in significant increase (P<0.05) in aortic diameter , intima-media thickness, PI, RI, renal artery PSV, PI, RI, intra-renal artery PSV, PI, and RI in comparison to the normal control group. There was no significant change (P>0.05) in aortic PSV and EDV, and no significant change (P>0.05) in renal artery and intra-renal artery EDV. These results may be explained as that the end diastolic velocity is not necessarily increased in stenosed arteries (normal or even low end diastolic velocity does not exclude critical vascular stenosis). EDV may be increased only in high degree of stenosis(201). 4.6.2 Effect of Sildenafil and Vardenafil treatment on end diastolic blood flow velocity Treatment of rabbits with sildenafil and vardenafil did not result in a significant change (P>0.05) in aortic EDV, but there was a significant decrease (P<0.05) in the renal artery and intra-renal artery EDV in comparison to that of the induced untreated group. This finding is consistent with that reported by Ercan et al. (2005)(198) who concluded that Sildenafil citrate had no significant effect on aortic and SMA circulation and only caused mild changes in the carotid artery circulation. This finding also explained by Ardicoglu et al. (2005)(199) who demonstrated that peroral sildenafil citrate usage had slight effects on hemodynamic parameters of lower segmental branch of right renal artery(199). 4.7 Effect on resistive index (RI) 4.7.1 Effect of cholesterol enriched diet on resistive index There was a significant increase (P<0.05) in aortic, renal artery and intra-renal arteries blood flow resistive index (RI) in rabbits fed with cholesterol enriched diet as compared to that of the normal control. This finding is consistent with Mohammad (2008)(77) who found that cholesterol enriched diet results in significant increase (P<0.05) in aortic diameter , intima-media thickness, PI, RI, renal artery PSV, PI, RI, intra-renal artery PSV, PI, and RI in comparison to the normal control group. This result can be explained by the fact that resistive index (RI) is a measure of resistance in the circulation and an increase in resistive index associated with arterial stiffness (which reflects atherosclerosis). Therefore, increased RI is associated with the severity of systemic atherosclerosis (137). 4.7.2 Effect of Sildenafil and Vardenafil treatment on resistive index In sildenafil treated and vardenafil treated rabbits there was non significant decrease (p>0.05) in aortic blood flow RI but a significant decrease (P<0.05) in renal artery and intra-renal arteries blood flow RI in comparison to that in the induced untreated group. This finding is consistent with (Lepore et al.) in patients with pulmonary hypertension are of particular interest . Sildenafil produced acute reductions in pulmonary artery pressure and pulmonary vascular resistance(87). These result are in agreement with Ercan et al. (2005)(198). Furthermore this finding in agreement with Enrique (2009)(200) who found that sildenafil increase significantly renal vascular flow and significantly decrease renal vascular resistance. The increased RI is associated with arterial stiffness and severity of atherosclerosis. So the treatment of rabbits with sildenafil or vardenafil results in reduction of RI. This is due to the fact that treatment with PDEIs results in reduction in the severity of atherosclerosis . This action on wave reflection properties implies that sildenafil reduces arterial pulse wave velocity and causes the reflected wave to be delayed in returning to the heart. This delay in arrival of the reflected wave reduces its amplitude, decreases systolic and pulse pressure, and reduces LV after load and myocardial oxygen demand. Because vasoactive drugs have little direct effect on large elastic arteries, the wave reflection action of sildenafil is likely confined to the muscular arteries.(202 ). 4.8 Effect on pulsatality index (PI) 4.8.1 Effect of cholesterol enriched diet on pulsatality index There was a significant increases (P<0.05) in aortic, renal artery and intra-renal arteries blood flow pulsatality indices in rabbits fed with cholesterol enriched diet as compared to that in the normal control. This finding is consistent with Mohammad (2008)(77) who found that cholesterol enriched diet results in significant increase (P<0.05) in aortic diameter , intima-media thickness, PI, RI, renal artery PSV, PI, RI, intra-renal artery PSV, PI, and RI in comparison to the normal control group. This result can be explained by the fact that pulsatality indices (PI) is a parameter closely related to the resistive index (RI). It has been shown to be related to vascular resistance. An increase in pulsatality index is associated with arterial stiffness and atherosclerosis(203) . 4.8.2 Effect of Sildenafil and Vardenafil treatment on pulsatality index In sildenafil treated and vardenafil treated rabbits, there was non significant decrease (p>0.05) in aortic blood flow pulsatality index but significant decrease (P<0.05) in renal artery and intra-renal arteries blood flow pulsatality indices in comparison to that in the induced untreated group. This finding is consistent with (Lepore et al.) (87).As well these result in agreement with Ercan et al. (2005)(198). The increased PI is associated with arterial stiffness and severity of atherosclerosis, therefore treatment of rabbits with sildenafil or vardenafil results in reduction in PI. This is due to the fact that treatment with PDEIs results in reduction in the severity of atherosclerosis . This action on wave reflection properties implies that sildenafil reduces arterial pulse wave velocity and causes the reflected wave to be delayed in returning to the heart. This delay in arrival of the reflected wave reduces its amplitude, decreases systolic and pulse pressure, and reduces LV after load and myocardial oxygen demand. Because vasoactive drugs have little direct effect on large elastic arteries, the wave reflection action of sildenafil is likely confined to the muscular arteries.(202 ). 4.9 Effect on renal artery-to-aortic peak systolic velocity ratio (RAR) 4.9.1 Effect of cholesterol enriched diet on renal artery–to-aortic peak systolic velocity ratio (RAR) There was significant increase (P<0.05) in renal artery –to-aortic peak systolic velocity ratio (RAR) in rabbits fed with cholesterol enriched diet as compared to the control of the same group. The explanation of this runs as follows: because there was a significant increase in the PSV of renal artery and non significant rise in the PSV of aorta, a significant increase in the RAR takes place which is an important Doppler index used in the diagnosis of renal artery stenosis (130). 4.9.2 Effect of Sildenafil and Vardenafil treatment on renal artery –toaortic peak systolic velocity ratio (RAR) In sildenafil treated and vardenafil treated rabbits, there was non significant decrease (p>0.05) in renal artery –to- aortic peak systolic velocity ratio in comparison to that in the induced untreated group. The reduction of RAR observed by vardenafil and sildenafil treatment was because the treatment with both drugs leads to a significant reduction in the PSV of renal artery but no significant reduction in the PSV of aorta. The reduction in RAR was not significant perhaps due to high cholesterol diet continued with the treatment which prevents the optimal action of the drugs or perhaps due to small sample size studied. Another reason for the non significant reduction in RAR may be due to the low doses of drugs and the short duration of treatment. 4.10 Effect on aortic histological change 4.10.1 Effect of cholesterol enriched diet on aortic histology All rabbits (100%) fed with cholesterol enriched diet had an involvement of their aorta with different phases of atherosclerotic lesions at the end of 12 weeks of the study that was significantly different (P<0.05) as compared with the normal control group. This finding is in agreement with that reported by Geetha et al. (2007)(204) and Asgary et al. (2007)(205) these finding might be due to atherogenic diet which encouraged atherosclerotic changes on aortic wall. 4.10.2 Effect of histology Sildenafil and Vardenafil treatment on aortic The overall histopathological features of rabbits treated with sildenafil and verdenafil are better than that of induced untreated group but this differences in severity of atherosclerotic changes dose not reach significant level (P>0.05). This finding may be due to the fact that rabbits were continued to be fed with cholesterol enriched diet together with the treatment with sildenafil and verdenafil which may prevent the maximal effect of the drugs or due to low doses of drugs and short duration of treatment. although sildenafil and vardenafil have the ability to improve endothelial function (206), and possess antioxidant action(186), additionally sildenafil might have an antiangiogenic effect, since sildenafil, tadalafil, and vardenafil have a beneficial effect on inflammatory activation and surrogate markers of endothelial dysfunction(206). 5.1 Conclusions The following points can be concluded out of this study: 1. High cholesterol diet causes a significant increase in aortic diameter, intima-media thickness, RI and PI and a significant increase in renal artery and intra-renal arteries blood flow PSV, PI and RI in male rabbits. 2. Sildenafil and Vardenafil did not significantly change the serum level of TC, TG, LDL-C, VLDL-C, HDL-C and the level atherogenic index in treated animals. 3. Both drugs significantly reduce the serum level of MDA and increase the serum level of GSH. 4. Both drugs did not significantly change the aortic intima-media thickness and aortic diameter of treated animals. 5. Both drugs significantly increase the arterial elasticity and decrease arterial stiffness as indicated by reduction of RI and PI of the aorta, and PSV,EDV,RI,PI of renal artery and intra-renal arteries. 5.2 Recommendations 1. In view of the effect of vardenafil and sildenafil in reducing of arterial resistance and stiffness as indicated by their ability to decrease PSV, PI and RI and have anti-oxidant effect. So further experimental studies with measurement of effect of vardenafil and sildenafil on inflammatory markers and cytokines which have major role in pathogenesis of atherosclerosis. 2. Because vardenafil and sildenafil treatment results in significant reduction in renal artery peak systolic velocity and non significant reduction in renal artery –to-aortic peak systolic velocity ratio which are Doppler indices used in the diagnosis of renal artery stenosis. So further experimental studies with large sample size and long periods about the effectiveness of vardenafil and sildenafil in treatment of renal artery stenosis may be required . 3. Different doses of vardenafil and sildenafil and combination with other PDEIs essential to reveal their effects on progression of atherosclerosis. 1. Anderson T. J.: Assessment and treatment of endothelial dysfunction in humans. J Am Coll Cardiol. 1999; 34(3):631-638. 2. Rosamond W. D., Chambless L.E., Folsom A.R., et al.: Trends in the incidence of myocardial infarction and in mortality due to coronary heart disease, 1998; 1987 to 1994. N Engl J Med; 339: 861-867. 3. Jules Y.T. L.: Heart and Blood Vessel Disorder. Atherosclerosis; 2008; www.merck.com. 4. Merck and Co. Inc.: Merck Manual of Geriatrics. Section 11, Cardiovascular Disorder, 2008; Chapter 83, Atherosclerosis; www.merck.com. 5. Fearon I. M. and Faux S. P.: Oxidative stress and cardiovascular disease: Novel tools give (free) radical insight, 2009; Journal of Molecular and Cellular Cardiology, YJMCC-06548; pages: 10; 4C: 6. Stocker R. and J. F. Keaney J. F.: Role of Oxidative Modifications in Atherosclerosis: Physiol. Rev.,2004; 84: 1381-1478. 7. Pyorala K., Laakso M., and Uusitupa M.: Diabetes and atherosclerosis: an epidemiologic view. Diabetes Metab Rev, 1987; 3: 463–524. 8. Jonasson L., Holm J., Skall O., Bondjers G., and Hansson G.K.: Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis,1986; 6: 131–138. 9. Vijver LP, Steyger R., Poppel G., Boer J.M., Kruijssen D.A., Seidell J.C., and Princen H.M.: Autoantibodies against MDA-LDL in subjects with severe and minor atherosclerosis and healthy population controls. Atherosclerosis,1996; 122: 245–253 10.Ross R. and Glomset J. A. : The pathogenesis of atherosclerosis. N Engl J Med, 1976; 295: 369–377. 11.Ross R. and Glomset J.A.: The pathogenesis of atherosclerosis. N Engl J Med , 1976; 295: 420–425. 12.Ross R.: Atherosclerosis—an inflammatory disease. N Engl J Med, 1999; 340: 115–126. 13.PFEM N., Fogelman A.M., Mottino G., and Frank J.S.: Lipid accumulation in rabbit aortic intima 2 hours after bolus infusion of low density lipoprotein. A deep-etch and immunolocalization study of ultrarapidly frozen tissue. Arterioscl Thromb, 1991; 11: 1795–1805. 14.Simionescu M. and Simionescu N.: Proatherosclerotic events: pathobiochemical changes occurring in the arterial wall before monocyte migration. FASEB J,1993; 7: 1359–1366. 15.Zilversmit D.B.: A proposal linking atherogenesis to the interaction of endothelial lipoprotein lipase with triglyceride-rich lipoproteins. Circ Res, 1973; 33: 633–638. 16.Camejo G., Fager G., Rosengren B., Camejo E. H., and Bondjers G.: Binding of low density lipoproteins by proteoglycans synthesized by proliferating and quiescent human arterial smooth muscle cells. J Biol Chem, 1993; 268: 14131–14137. 17.Borén J., Olin K., Lee I., Chait A., Wight T.N., and Innerarity T.L.: Identification of the principal proteoglycan-binding site in LDL. A singlepoint mutation in apo-B100 severely affects proteoglycan interaction without affecting LDL receptor binding. J Clin Invest, 1998; 101: 2658– 2664. 18.Skalen K., Gustafsson M., Rydberg E.K., Hulten L.M., Wiklund O., Innerarity T.L., and Borén J.: Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature, 2002; 417: 750–754. 19.Williams K.J., Petrie K.A., Brocia R.W., and Swenson T.L.: Lipoprotein lipase modulates net secretory output of apolipoprotein B in vitro. A possible pathophysiologic explanation for familial combined hyperlipidemia. J Clin Invest, 1991; 88: 1300–1306. 20.Williams K.J., Fless G.M., Petrie K.A., Snyder M.L., Brocia R.W., and Swenson T.L.: Mechanisms by which lipoprotein lipase alters cellular metabolism of lipoprotein(a), low density lipoprotein, and nascent lipoproteins. Roles for low density lipoprotein receptors and heparan sulfate proteoglycans. J Biol Chem, 1992; 267: 13284–13292. 21.Tamminen M., Mottino G., Qiao J.H., Breslow J.L., and Frank J.S.: Ultrastructure of early lipid accumulation in ApoE-deficient mice. Arterioscler Thromb Vasc Biol, 1999; 19: 847–853. 22.Hakala J.K., Oksjoki R., Laine P., Du H., Grabowski G.A., Kovanen P.T., and Pentikäinen M.O.: Lysosomal enzymes are released from cultured human macrophages, hydrolyze LDL in vitro, and are present extracellularly in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol, 2003; 23: 1430–1436. 23.Ismail N.A., Alavi M.Z., and Moore S.: Lipoprotein-proteoglycan complexes from injured rabbit aortas accelerate lipoprotein uptake by arterial smooth muscle cells. Atherosclerosis, 1994; 105: 79–87. 24.Vijayagopal P., Srinivasan S.R., Radhakrishnamurthy B., and Berenson G.S. ): Lipoprotein-proteoglycan complexes from atherosclerotic lesions promote cholesteryl ester accumulation in human monocytes/macrophages. Arterioscl Thromb, 1992; 12: 237–249. 25.Goldstein J.L., Ho Y.K., Basu S.K., and Brown M.S.: Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc Natl Acad Sci USA, 1979; 76: 333–337. 26.Henriksen T., Mahoney E.M., and Steinberg D.: Enhanced macrophage degradation of low density lipoprotein previously incubated with cultured endothelial cells: recognition by receptors for acetylated low density lipoproteins. Proc Natl Acad Sci USA, 1981; 78: 6499–6503. 27.Steinbrecher U.P., Parthasarathy S., Leake D.S., Witztum J.L., and Steinberg D.: Modification of low density lipoprotein by endothelial cells involves lipid peroxidation and degradation of low density lipoprotein phospholipids. Proc Natl Acad Sci USA, 1984; 81: 3883–3887. 28.Haberland M.E., Fogelman A.M., and Edwards P.A.: Specificity of receptor-mediated recognition of malonydialdehyde-modified low density lipoproteins. Proc Natl Acad Sci USA, 1982; 79: 1712–1716. 29.Haberland M.E., Olch C.L., and Fogelman A.M.: Role of lysines in mediating interaction of modified low density lipoproteins with the scavenger receptor of human monocyte macrophages. J Biol Chem, 1984; 259: 11305–11311. 30.Cushing S.D., Berliner J.A., Valente A.J., Territo M.C., Navab M., Parhami F., Gerrity R., Schwartz C.J., and Fogelman A.M.: Minimally modified low density lipoprotein induces monocyte chemotactic protein (MCP-1) in human endothelial and smooth muscle cells. Proc Natl Acad Sci USA, 1990; 87: 5134–5138. 31.Rajavashisth T.B., Andalibi A., Territo M.C., Berliner J.A., Navab M., Fogelman A.M., and Lusis A.J.: Induction of endothelial cell expression of granulocyte and macrophage colony-stimulating factors by modified low-density lipoproteins. Nature, 1990; 344: 254–257. 32.Navab M., Imes S.S., Hama S.Y., Hough G.P., Ross L.A., Bork R.W., Valente A.J., Berliner J.A., Drinkwater D.C., Laks H., and Fogelman A.M.: Monocyte transmigration induced by modification of low density lipoprotein in cocultures of human aortic wall cells is due to induction of monocyte chemotactic protein 1 synthesis and is abolished by high density lipoprotein. J Clin Invest, 1991 88: 2039–2046. 33.Boring L., Gosling J., Cleary M., and Charo I.F.: Decreased lesion formation in CCR2–/– mice reveals a role for chemokines in the initiation of atherosclerosis. Nature, 1998; 394: 894–897. 34.Gosling J., Slaymaker S., Gu L., Tseng S., Zlot C.H., Young S.G., Rollins B.J., and Charo I.F.: MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. J Clin Invest, 1999; 103: 773–778. 35.Quinn M.T., Parthasarathy S., Fong L.G., and Steinberg D.: Oxidatively modified low density lipoproteins: a potential role in recruitment and retention of monocyte/macrophages during atherogenesis. Proc Natl Acad Sci USA, 1987; 84: 2995–2998. 36.McMurray H.F., Parthasarathy S., and Steinberg D.: Oxidatively modified low density lipoprotein is a chemoattractant for human T lymphocytes. J Clin Invest, 1993; 92: 1004–1008. 37.Rahm A.S., Nilsson A.H., Regnstrom J., Hamsten A., and Nilsson J.: Native and oxidized LDL enhances production of PDGF AA and the surface expression of PDGF receptors in cultured human smooth muscle cells. Arterioscler Thromb, 1992; 12: 1099–1109. 38.Parums D.V., Brown D.L., and Mitchinson M.J.: Serum antibodies to oxidized low-density lipoprotein and ceroid in chronic periaortitis. Arch Pathol Lab Med, 1990; 114: 383–387. 39.Salonen J.T., Herttuala S. Y., Yamamoto R., Butler S., Korpela H., Salonen R., Nyyssönen K., Palinski W., and Witztum J.L.: Autoantibody against oxidised LDL and progression of carotid atherosclerosis. Lancet, 1992; 339: 883–887. 40.Griffith R.L., Virella G.T., Stevenson H.C., and Virella M.F.L.: Low density lipoprotein metabolism by human macrophages activated with low density lipoprotein immune complexes A possible mechanism of foam cell formation. J Exp Med, 1988; 168: 1041–1059. 41.Klimov A.N., Denisenko A.D., Popov A.V., Nagornev V.A., Pleskov V.M., Vinogradov A.G., Denisenko T.V., Magracheva E., Kheifes G.M., and Kuznetzov A.S.: Lipoprotein-antibody immune complexes. Their catabolism and role in foam cell formation. Atherosclerosis, 1985; 58: 1– 15. 42.Iuchi T., Akaike M., Mitsui T., Ohshima Y., Shintani Y., Azuma H., and Matsumoto T.: Glucocorticoid excess induces superoxide production in vascular endothelial cells and elicits vascular endothelial dysfunction. Circ Res., 2003; 92:81–7. 43.Madamanchi N.R., and Runge M.S.: Mitochondrial dysfunction in atherosclerosis. Circ Res:, 2007; 100: 460–73. 44.Valko M., Izakovic M., Mazur M., Rhodes C.J., and Telser J.: Role of oxygen radicals in DNA damage and cancer incidence, Mol. Cell. Biochem, 2004; 266: 37–56. 45.Chen J., and Mehta J.L. () : Role-of-Oxidative-Stress-in-Coronary-HeartDisease. IHJ., Apr. 2004; 56: 1-17. 46.Galle J., and Heermeier K.: Angiotensin II and oxidized LDL: an unholy alliance creating oxidative stress. Nephrol Dial Transplant, 1999; 14: 2585–2589. 47.Nicholls D.G., and Budd S.L.: Mitochondria and neuronal survival. Physiol Rev., 2000; 80: 315–360. 48.Inoue M., Sato E.F., Nishikawa M., Park A.M., Kira Y., Imada I., and Utsumi K.: Mitochondrial generation of reactive oxygen species and its role in aerobic life, Curr. Med. Chem., 2003; 10: 2495–2505. 49.Cadenas E., and Davies K.J.A.: Mitochondrial free radical generation, oxidative stress, and aging, Free Rad. Biol. Med., 2000; 29: 222–230. 50.Li C.Y., and Jackson R.M.: Reactive species mechanisms of cellular hypoxia-reoxygenation injury, Am. J. Physiol.-Cell Physiol, 2002; 282 C227–C241. 51.Conner E.M.,: Grisham, Inflammation, free radicals, and antioxidants, Nutrition, 1996; 12: 274–277. 52.Hayes J.D., and McLellan L.I.: Glutathione and glutathionedependentenzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic Res., 1999; 31: 273–300. 53.Warnholtz A., Nickenig G., Schulz E., Macharzina R., Brasen J.H., Skatchkov M., et al.: Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: evidence for involvement of the renin-angiotensin system. Circulation, 1999; 99: 2027–2033. 54.Berliner J.A., and Heinecke J.W.: The role of oxidized lipoproteins in atherogenesis. Free Radic Biol Med., 1996; 20: 707–727. 55.Heinecke J.W.: Oxidants and antioxidants in the pathogenesis of atherosclerosis: implications for the oxidized low-density lipoprotein hypothesis. Atherosclerosis, 1998; 141: 1–15. 56.Weinbrenner T., Cladellas M., Covas M. I., Fito M., Tomas M., Senti M., et al.: High oxidative stress in patients with stable coronary heart disease. Atherosclerosis, 2003; 168: 99–106. 57.Li D., Chen H., Romeo F., Sawamura T., Saldeen T., and Mehta J.L.: Statins modulate oxidized low-density lipoprotein-mediated adhesion molecule expression in human coronary artery endothelial cells: role of LOX-1. J Pharmacol Exp Ther., 2002; 302: 601–605. 58.Rosenson R.S., and Brown A.S.: Statin use in acute coronary syndromes: cellular mechanisms and clinical evidence. Curr Opin Lipidol, 2002; 13: 625–630. 59.Ananyeva N.M., Tjurmin A.V., Berliner J.A., Chisolm G.M., Liau G., Winkles J.A., et al.: Oxidized LDL mediates the release of fibroblast growth factor-1. Arterioscler Thromb Vasc Biol., 1997; 17: 445–453. 60.Li D., Liu L., Chen H., Sawamura T., Ranganathan S., and Mehta J.L.: LOX-1 mediates oxidized low-density lipoprotein-induced expression of matrix metalloproteinases in human coronary artery endothelial cells. Circulation, 2003; 107:612–617. 61.Pepine C.J., Drexler H., and Dzau V.J.: Endothelial function in cardiovascular health and disease. New York, Landmark Programs, 1997. 62.Luscher T.F., and Barton M.: Biology of the endothelium, Clin. Cardiol, 1997; 20: 3–10. 63.Drexler H.: Endothelial dysfunction: clinical implications, Prog. Cardiovasc. Dis., 1997; 39: 287–324. 64.Celermajer D.S.: Endothelial dysfunction: does it matter? Is it reversible?, J. Am. Coll. Cardiol, 1997; 30: 325–333. 65.Cooke J.P., and Dzau V.J.: Nitric oxide synthase: role in the genesis of vascular disease, Annu. Rev. Med., 1997; 48: 489–509. 66.Wever R.M., Luscher T.F., Cosentino F., and Rabelink T.J.: Atherosclerosis and the two faces of endothelial nitric oxide synthase, Circulation, 1998; 97: 108–112. 67.Matsuoka H.: Endothelial dysfunction associated with oxidative stress in human. Diabetes Research and Clinical Practice 54 Suppl., 2001; 2: S65– S72. 68.Boger R. H., Boger S.M.B., Kienke S., Stan A.C., Nafe R., and Frolich J.C.: Dietary L-arginine decreases myointimal cell proliferation and vascular monocyte accumulation in cholesterol-fed rabbits. Atherosclerosis, 1998; 136: 67-77. 69.Bult H.: Nitric oxide and atherosclerosis: possible implications for therapy. Mol Med Today, 1996; 2: 510-518. 70.Rubbo H, Batthyany C. and Radi R.: Nitric Oxide - Oxygen Radicals Interactions in Atherosclerosis. Biol. Res., 2000; 33: 1-8. 71.Rubbo H., Radi R., Trujillo M., Telleri R., Kalyanaraman B., Barnes S., Kirk M., and Freeman B.A.: Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogencontaining oxidized lipid derivatives. J Biol Chem., 1994; 269: 2606626075. 72.Rubbo H., Radi R., Anselmi D., Kirk M., Barnes S., Butler J., Eiserich J.P., and Freeman B.A.: Nitric oxide reaction with lipid peroxyl radicals spares alpha-tocopherol during lipid peroxidation. Greater oxidant protection from the pair nitric oxide/alpha-tocopherol than alphatocopherol/ascorbate. J Biol Chem., 2000; 275: 10812-10818. 73.Busse R., and Fleming I.: Endothelial dysfunction in atherosclerosis. J Vasc Res., 1996; 33: 181-194. 74.Edwards J. W. R., Manson J. E., Hennekens C. H., and Buring J. E.: The Primary Prevention of Coronary Heart Disease in Women. N Engl J Med., 1995; 332: 1758-1766. 75.Kreisberg R. A., and Oberman A.: Medical Management of Hyperlipidemia /Dyslipidemia. J Clin Endocrinol Metab., 2003; 88: 2445-2461. 76.Esselstyn C.B.: Resolving the Coronary Artery Disease Epidemic through Plant-Based Nutrition, 2001. 77.Younis M. A. M.: A study of the effects of statins on aortic intima media thickness and blood flow velocities of aorta and renal artery in hyperlipidemic male rabbits. M.Sc. Thesis, College of Medicine, Kufa University, 2008. 78.Lakka T. A.; Salonen R., Kaplan G. A., and Salonen J. T.: Blood Pressure and the Progression of Carotid Atherosclerosis in Middle-Aged Men . Hypertension. 1999; 34: 51-56. 79.Kreisberg R.A., Oberman A.: Lipids and atherosclerosis: lessons learned from randomized controlled trials of lipid lowering and other relevant studies. J Clin Endocrinol Metab., 2002 87: 423–437. 80.Kharbanda R. and MacAllister R.J.: The Atherosclerosis Time-Line and the Role of the Endothelium, 2005; Curr. Med. Chem. – Immun., Endoc. & Metab. Agents, 5, 47-52. 81.DiscoveRx, Name, Logo, and Hunter : Measurement of Phosphodiesterase Activity using HitHunter cAMP, 2004; II. 1–3. 82.Cheitlin M.D., Hutter A.M., Brindis R.G., Ganz P., Kaul S., Russell R.O., et. al.: Use of Sildenafil (Viagra) in Patients With Cardiovascular Disease. Circulation, 1999; 99: 168-177. 83.Terrett N.K.: Sildenafil (Viagra), a potent and selective inhibitor of Type 5 cGMP phosphodiesterase with utility for the treatment of male erectile dysfunction. Bioorg Med Chem Lett., 1996; 6: 1819–1824. doi:10.1016/0960-894X(96)00323-X. 84.Kling J.: From hypertension to angina to Viagra" ([dead link] – Scholar search). Mod Drug Discov., 1998 1: 31–38. http://pubs.acs.org/hotartcl/mdd/98/novdec/viagra.html. 85.Lau L.C., Adaikan P.G.: Mechanisms of direct relaxant effect of sildenafil, tadalafil and vardenafil on corpus cavernosum. Eur J Pharmacol, Jul. 2006; 541(3):184-190. 86.Tessier D.J., Komalavilas P., McLemore E., Thresher J., and Brophy C.M.: Sildenafil- induced vasorelaxation is associated with increases in the phosphorylation of the heat shock-related protein 20 (HSP20).Journal of Surgical Reaserch, 2004; 118 (1): 21-25. 87.Gillies H.C., Roblin D., and Jackson G.: Coronary and systemic hemodynamic effects of sildenafil citrate: from basic science to clinical studies in patients with cardiovascular disease. International Journal of Cardiology, 2002; 86: 131–141. 88.Park J.W., Mrowietz C., Chung N., and Jung F.: Sildenafil improves cutaneous microcirculation in patients with coronary artery disease: a monocentric, prospective, double-blind, placebo-controlled, randomized cross-over study. Clin Hemorheol Microcirc, 2004; 31(3):173-183. 89.Fries R., Shariat K., Wilmowsky H. V, and Böhm M.: Sildenafil in the Treatment of Raynaud’s Phenomenon Resistant to Vasodilatory Therapy. Circulation, 2005; 112: 2980-2985. 90.Jackson G., Montorsi P., and Cheitlin M.D.: Cardiovascular safety of sildenafil citrate (Viagra): an updated perspective. Urology, 2006; 68(3): 47-60. 91.Ishikura F., Beppu S., Hamada T., Khandheria B.K., Seward J.B., and Nehra A.: Effects of Sildenafil Citrate (Viagra) Combined With Nit rate on the Heart. Circulation, 2000; 102: 2516-2553. 92.Bhalla A., Singh R., Gautam C.S., and Sachdev A.: Sildenafil Citrate In Diabetics With Hypertension And Erectile Dysfunction. The Internet Journal of Internal Medicine, 2003; 1528-8382. 93.Gross G.J.: Sildenafil and Endothelial Dysfunction in Humans. Circulation, 2005; 111: 721-723. 94.Piccirillo G., Nocco M., Lionetti M., Moise A., Naso C., Marigliano V., and Cacciafesta M.: Effects of sildenafil citrate (Viagra) on cardiac repolarization and on autonomic control in subjects with chronic heart failure. The American heart journal, 2002; 143(4): 703-710 . 95.Kaya D. , Guler C., Metin E.A., Barutcu I. , and Dincel C.: Sildenafil citrate does not alter ventricular repolarization properties: Novel evidence from dynamic QT analysis. Annals of noninvasive electrocardiology, 2004 9(3): 228-23. 96.Swissa M., Ohara T., Lee M.H., Kaul S., Shah P.K., Hayashi H., et. al.: Sildenafil-nitric oxide donor combination promotes ventricular tachyarrhythmias in the swine right ventricle. Am J Physiol Heart Circ Physiol, 2002; 282: 1787-1792. 97.Phillips B.G., Kato M., Pesek C.A., Winnicki M., Narkiewicz K., Davison D., et. al.: Sympathetic Activation by Sildenafil. Circulation, 2000; 102: 3068-3095. 98.Borlaug B.A., Melenovsky V., Marhin T., Marhin T., Fitzgerald P., and Kass D.A.: Sildenafil inhibits beta-adrenergic-stimulated cardiac contractility in humans. Circulation, 2005; 25; 112 (17):2642-2649. 99.Patrizi R., Leonardo F., Pelliccia F., Chierchia S.L., Galetta P., Cerquetani E., et. al.: Effect of sildenafil citrate upon myocardial ischemia in patients with chronic stable angina in therapy with beta-blockers. Ital Heart J., 2-001; 2: 841-843. 100. Melo S.E. F., Toledo J.C.Y., Coelho O.R., Luca I.M.D., Santos J.E.T, Hyslop S., et. al.: Sildenafil reduces cardiovascular remodeling associated with hypertensive cardiomyopathy in NOS inhibitor-treated rats. Eur J Pharmacol, 2006; 542(1-3):141-147. 101. Hassan M.A.H., and Ketat A.F.: Sildenafil citrate increases myocardial cGMP content in rat heart, decreases its hypertrophic response to isoproterenol and decreases myocardial leak of creatine kinase and troponin T. BMC Pharmacol, 2005; 5: 10-23. 102. Fung E., Fiscus R.R., Yim A.P.C., Angelini G.D., and Arifi A.A.: The Potential Use of Type-5 Phosphodiesterase Inhibitors in Coronary Artery Bypass Graft Surgery. Chest., 2005; 128: 3065-3073. 103. Highleyman L.: Protease inhibitors and sildenafil (Viagra) should not be combined. BETA., 1999; 12(2): 3-9. 104. Bischoff E.: Potency, selectivity, and consequences of nonselectivity of PDE inhibition. International journal of impotence research. PDE-5 Inhibitor Therapy for Erectile Dysfunction, 2004; 16(1): 11-14. 105. Eardley I.: Vardenafil: a new oral treatment for erectile dysfunction. Int J Clin Pract., 2004; 58(8): 801-806. 106. www.fda-gov/bbs/topics/NEWS/2007/NEW01730.html 107. Ghofrani H.A., Voswinckel R., Reichenberger F., et al.: Differences in hemodynamic and oxygenation responses to three different phosphodiesterase-5 inhibitors in patients with pulmonary arterial hypertension. A randomized prospective study. J Am Coll Cardiol, 2004; 44:1488–96. 108. Thadani U., Smith W., Nash S., et al.: The effect of vardenafil, a potent and highly selective phosphodiesterase-5 inhibitor for the treatment of erectile dysfunction, on the cardiovascular response to exercise in patients with coronary artery disease. J Am Coll Cardiol., 2002; 40(11). 109. Pomara G., Morelli G., Pomara S., et al.: Cardiovascular parameter changes in patients with erectile dysfunction using pde-5 inhibitors: a study with sildenafil and vardenafil. J Androl, 2004; 25(4): 625-629. 110. Kendirci M., Bivalacqua T.J., and Hellstrom W.J.G.: Vardenafil: a novel type 5 phosphodiesterase inhibitor for the treatment of erectile dysfunction. Expert Opinion on Pharmacotherapy, 2004; 5(4): 923-932. 111. Xin Z.: Cardiovascular safety of vardenafil. Zhonghua Nan Ke Xue Journal, 2004; 10(10): 790-793. 112. Wespes E., Amar E., Hatzichristou D., Hatzimouratidis K., Montorsi F., Pryor J., and Vardi Y.: Guidelines on Erectil Dys Function, European Association of Urology, 2005; pp. 27. 113. Clemmesen J.O., Giraldi A., Ott P., Dalhoff K., Hansen B.A., and Larsen F. S.: Sildenafil does not influence hepatic venous pressure gradient in patients with cirrhosis World J Gastroenterol, 2008; 28; 14(40): 62086212. 114. Szabo G., Radovits T., Veres G., Krieger N., Loganathan S., Sandner P., and Karck M.: ardenafil protects against myocardial and endothelial injuries after cardiopulmonary bypass. European Journal of Cardio-thoracic Surgery, 2009; 36: 657—664. 115. Salloum F.N., Ockaili R.A., Wittkamp M., Marwaha V.R.,and Kukreja R.C.: Vardenafil: a novel type 5 phosphodiesterase inhibitor reduces myocardial infarct size following ischemia/reperfusion injury via opening of mitochondrial K(ATP) channels in rabbits. J Mol Cell Cardiol, 2006; 40(3): 405-411. 116. Brand M., and Deussen A. : Vardenafil increases coronary flow response to hypercapnic acidosis in isolated guinea pig heart. Basic Res Cardiol, 2006; pp. 27. 117. Billups K. L.: Erectile dysfunction as an early indicator of cardiovascular and metabolic disease. Heart Metab, 2005; 28:5–9. 118. Gupta M., Kovar A., and Meibohm B.: The Clinical Pharmacokinetics of Phosphodiesterase-5 Inhibitors for Erectile Dysfunction. J. Clin. Pharmacol, 2005; 45: 987-1002. 119. Reffelmann T., and Kloner R.A.: Therapeutic Potential of Phosphodiesterase 5 Inhibition for Cardiovascular Disease. Circulation, 2003; 108: 239-250. 120. Kloner R.A.: Cardiovascular Effects of the 3 Phosphodiesterase-5 Inhibitors Approved for the Treatment of Erectile Dysfunction. Circulation, 2004; 110: 3149-3155. 121. Briganti A., Salonia A., Deho F., Zanni G., Barbieri L., Rigatti P., et al.: Clinical update on phosphodiesterase type-5 inhibitors for erectile dysfunction. World Journal of Urology, 2005. 122. Veloso H.H., and Paola A.A.V.: Atrial fibrillation after vardenafil therapy. Emergency Medicine Journal, 2005; 22:823-829. 123. Russell S.t., Khandheria B.k., and Nehra A.: Erectile Dysfunction and Cardiovascular Disease. Mayo Clin Proc., 2004; 79:782-794. 124. Auerbach S.M., Gittelman M., Mazzu A., Cihon F., Sundaresan P., and White W.B.: Simultaneous administration of vardenafil and tamsulosin does not induce clinically significant hypotension in patients with benign prostatic hyperplasia. Urology, 2004; 64(5): 998-1003. 125. Leiner T., Kessels A. G. H., Nelemans P. J., et al.: Peripheral Arterial Disease: Comparison of Color Duplex US and Contrast-enhanced MR Angiography for Diagnosis. Radiology, 2005; 235: 699-708. 126. Sabeti S., Schillinger M., and Mlekusch W., et al.: Quantification of Internal Carotid Artery Stenosis with Duplex US: Comparative Analysis of Different Flow Velocity Criteria. Radiology, 2004; 232: 431-439. 127. Wetzner S. M., Kiser L. C., Bezreh J. S.: Duplex Ultrasound Imaging. Vascular Applications. Radiology, 1984; 150: 507-514. 128. Koelemay M.J., Hartog D., Prins M.H., Kromhout J.G., Legemate D.A., and Jacobs M.J.: Diagnosis of arterial disease of the lower extremities with duplex ultrasonography. Br J Surg., 1996; 83: 404-409. 129. Olin J. W.; Piedmonte M. R.; Young J.R.;. Anna S. D; Grubb M.; and Childs M. B.: The Utility of Duplex Ultrasound Scanning of the Renal Arteries for Diagnosing Significant Renal Artery Stenosis. Annal of internal medicine, 1995; Volume 122 (11): 833-838. 130. Dubbin P. : renal artery stenosis – duplex Doppler evaluation . British j. of radiology1986; 59: 225-229. 131. Petersen L. J., Petersen J. R., Talleruphuus U., Ladefoged S. D., Mehlsen J., and Jensen H .E.: The pulsatility index and the resistive index in renal arteries. Associations with long-term progression in chronic renal failure. Nephrol Dial Transplant, 1997; 12: 1376–1380. 132. Allan P.L., Dubbins P. A., Pozniak M. A., and Mc-Dicken W. N.: Clinical Doppler ultrasound, 1rst ed., 2002; chapter 6. page 114. 133. Gosling R.G., Dunbar G., King D.H. et al.: The quantitative analysis of occlusive peripheral arterial disease by non invasive ultrasonic technique . Angiology, 1971; 22: 52-55. 134. Petersen L.J ., Petersen J.R., Ladefoged S.D., Mehlsin J., and Jensen H.E.: The pulsatality index and the resistive index in renal arteries in patients with hypertension and chronic renal failure. Nephrol Dial Transplant, 1995; 10: 2060–2064. 135. Bardelli M., Jensen G., Volkmann R., and Aurell M.: Non-invasive ultrasound assessment of renal artery stenosis by means of the Gosling pulsatility index. J. Hypertens, 1992; 10: 985–989. 136. Hoffmann U., Edwards J.M., Carter S. et al.): Role of duplex scanning for the detection of atherosclerotic renal artery disease. Kidney Int., 1991; 39: 1232-1239. 137. Ohta Y., Fujii K., Arima H., et al.: Increased renal resistive index in atherosclerosis and diabetic nephropathy assessed by Doppler sonography. J Hypertens, 2005; 23(10): 1905-1911. 138. Ganong W. F.: review of medical physiology 20th ed., 2001; chapter 30 page 562. 139. Paut O. and Bissonnette B.: Effects of temperature and haematocrit on the relationships between blood flow velocity and blood flow in a vessel of fixed diameter. British Journal of Anaesthesia, 2002; 88: 277-279. 140. Sutera S. P., and Skalak R. : The history of Poiseuille's law. Annual Review of Fluid Mechanics, 1993; 25: 1-19. 141. Gruber E.M., Jonas R.A., Newburger J.W., et al.: The effect of hematocrit on cerebral blood flow velocity in neonates and infants undergoing deep hypothermic cardiopulmonary bypass. Anesth. Analg., 1999; 89: 322–327. 142. Rosenkrantz T., Stonestreet B., Hansen N., Nowicki P.: Cerebral blood flow in the newborn lamb with polycythemia and hyper viscosity. J. Pediatrics, 1984; 104: 276–80. 143. Eckmann D., Bowers S., Stecker M., and Cheung A.: Hematocrit, volume expander, temperature and shear rate effects on blood viscosity. Anesth Analg, 2000; 91: 539–45. 144. Nitta N., Homma K. , and Shiina T.: Pressure Gradient Estimation Based on Ultrasonic Blood Flow Measurement. Jpn. J. Appl. Phys., 2006; 45: 4740-4748. 145. Bortel L. M. V. ,and Spek J. J.: Influence of aging on arterial compliance. j of human hypertension., 1998; 12 : 583-586. 146. Stary H.C., Chandler A.B., Dinsmore R.E., et al.): A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis: a report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation, 1995; 92 : 1355-1374. 147. Otto C. M.: Valvular Heart Disease: second ed., 2004; chapter 3 page 65. 148. Yoganathan A.P.: Fluid mechanics of aortic stenosis . Eur Heart J, 1988; 9 (supple E) : 13-17. 149. Allan P. L., Dubbins P. A., Pozniak M. A. , and McDicken W. N.: Clinical Doppler ultrasound ,1rst ed., 2002; chapter 6, page 114. 150. De-Smet A.A., and Kitslaar P.J.: A duplex criteria for aortic stenosis , European. journal of vascular surgery, 1990; 4: 275-278. 151. Guyton A. C., and Hall J. E.: Textbook of medical physiology: 11th ed., 2006; chapter 26, page 321. 152. Gosset J., and Olin J. W.: Atherosclerotic Reno vascular Disease: Clinical Clues and Natural History. Journal of Endovascular Surgery, 1997; 4 (3): 316–320. 153. Allan P. L., Dubbins P. A.,. Pozniak M. A , and McDicken W. N.: Clinical Doppler ultrasound, 2002;1rst ed. chapter 8, page 174. 154. Zwain A. A. M. H., and Alshamma Y. M. H.: A non invasive evaluation of variability of intra-renal blood flow at rest and during exercise. Kufa Med. Journal, 2008; 11(1):348-364. 155. Ji H., Shen H., Uhanova J., Zhang M., Minuk G.Y., and Gong Y.: Effects of sildenafil citrate on hepatic function and regeneration in normal and alcohol-fed rats. Liver Int., 2005; 25(4):913-919. 156. AL- Hakeem A. H. M.: Study of the Effect of Phosphodiestrease-5Inhiitor on Some Coagulation Parameters, Lipid Profile and Histopathological Changes in the Eye of Male Rats. Ms. C. Thesis, College of Medicine, University of Kufa, 2007; pp. 129. 157. Hu M. Y., Li Y.L., Jiang C. H., Liu Z. Q., Qu S. L., and Huang Y. M.: Comparison of lycopene and fluvastatin effects on atherosclerosis induced by a high-fat diet in rabbits, 2008; Nutrition 24, 1030–1038. 158. Narayanasawamy M., Wright K. C., and Kandarpa K.: Animal Models for Atherosclerosis, Restenosis, and Endovascular Graft Research: J. vascular interventional radiology, 2000; 11: 5-17. 159. Bischoff E., Niewoehner U., Haning H., Sayed M. E., Schenke T., and Schlemmer K. H.:The Oral Efficacy Of Vardenafil Ydrochloride For Inducing Penile Erection In A Conscious Rabbit Model. The Journal Of Urology, 2001; 165, 1316–1318. 160. Mayne S.T.: Antioxidant nutrient and chronic disease: use of biomarker of exposure and oxidative stress status in epidemiologic research. J nutrition, 2003; 133;933-940. 161. Engelmann B.: Changes of membrane phospholipid composition of human erythrocytes in hyperlipidemias. Increased phosphatidyl -choline and reduced sphingomyelin in patients with elevated levels of triacylglycerol-rich lipoproteins. Biochim. Biophys. Acta., 1992; 1165: 32–37. 162. Ashwood E.R., and Saunders W.B.: TIETZ N. W. Text book of clinical chemistry, 1999; 3 rd ed. page 826-835. 163. Allian C. C.: Clin. Chem., 1974; 20:4 , pp.470-475. 164. Trinder P.: Ann. Clin. Biochem, 1969; 6, pp. 27-29. 165. Fossati P., and Prencipe L.: Clin. Chem., 1982; 28,pp.2077-2080. 166. Grove T. H.: Effect of reagent PH on determination of HDL cholesterol by precipitation with sodium phosphotungstate-magnesium. Clin Chem., 1979; 25:560. 167. Prasad K.: Hypocholesterolemic and antiatherosclerotic effect of flax lignan complex isolated from flaxseed. Atherosclerosis, 2005; 179:269– 75. 168. Hu M.L.: Measurement of protein thiol groups and glutathione in plasma. Methods Enzymol, 1994; 233: 381–5. 169. Quesenberry K.E., and Carpenter J.W.: Ferrets, Rabbits and Rodents: Clinical Medicine and Surgery, Saunders: St. Louis, 2004. 170. Victor P. M., Hulst V. D., Baalen J. V., and Kool L. S.: Renal Artery Stenosis. Endovascular Flow Wire Study for Validation of Doppler U.S. Radiology. 1996; 200:165-168. 171. Eicke B.M., Buss E., Bahr R.R., Hajak J. and Paulus W.: Influence of Acetozolamide and CO2 on Extracrinal Flow Volume and Intracrinal Blood Flow Velocity: J Strock, 1999; 30: 76-80. 172. Prasad K., and Lee P.: Suppression of oxidative stress as a mechanism of reduction of hypocholesterolemic atherosclerosis by aspirin. J Cardiovasc Pharmacol Therapeut, 2003; 8: 61–9. 173. Lange W. H.: Statistics techniques in business and economics. 2002; McGraw Hill/Irwin, eleventh edition, pp. 408. 174. Romero F., Iturbe B. R., Pons H., Parra G., Quiroz Y., and Gonza´lez J. R. L.: Mycophenolate mofetil treatment reduces cholesterol-induced atherosclerosis in the rabbit. Atherosclerosis, 2000; 152 : 127–133. 175. Romero F.: Mycophenolate mofetil treatment reduces cholesterolinduced atherosclerosis in the rabbit. Atherosclerosis, 2000;152: 127– 133. 176. Prasad K., and Lee P.: Suppression of hypercholesterolemic atherosclerosis by pentoxifylline and its mechanism. Atherosclerosis, 2007; 192: 313–22. 177. Nigrisa F. D., and Paolo F.: Therapeutic dose of nebivolol, a nitric oxide-releasing b-blocker, reduce atherosclerosis in cholesterol-fed rabbits. Nitric Oxide, 2008; 19 : 57–63. 178. Kimura M., Higashi Y., Hara K., Noma K., Sasaki S., Nakagawa K., Goto C., Oshima T., Yoshizumi M., and Chayama K.: PDE5 Inhibitor Sildenafil Citrate Augments Endothelium-Dependent Vasodilation in Smokers. American Heart Association, Inc., 2003; 41:1106-1110. 179. Balkan J., and Abbasog˘lu S. D.: The effect of a high cholesterol diet on lipids and oxidative stress in plasma, liver and aorta of rabbits and rats.Nutrition Research, 2004; 24: 229–234. 180. Prasad K., and Kalra J.: Oxygen free radicals and hypercholesterolemic atherosclerosis: effect of Vitamin E. Am Heart J, 1993;125: 958–73. 181. Prasad K.: Regression of hypercholesterolemic atherosclerosis in rabbits by secoisolariciresinol diglucoside isolated from flaxseed. Atherosclerosis, 2008; 197: 34–42. 182. Prasad K. : Reduction of serum cholesterol and hypercholesterolemic atherosclerosis in rabbits by secoisolariciresinol diglucoside isolated from flaxseed. Circulation, 1999; 99:1355–62. 183. Prasad K. : Hypocholesterolemic and antiatherosclerotic effect of flax lignan complex isolated from flaxseed. Atherosclerosis, 2005; 179: 269–75. 184. Prasad K, and Lee P. : Suppression of oxidative stress as a mechanism of reduction of hypocholesterolemic atherosclerosis by aspirin. J Cardiovasc Pharmacol Therapeut, 2003; 8:61–9. 185. Abdollahi M.: Protection by sildenafil and theophylline of lead acetate-induced oxidative stress in rat submandibular gland and saliva. Human & Experimental Toxicology, 2003; 22(11): 587-592. 186. Abdollahi M., Moghadam A. B., Emami B., Fooladian .F, and Zafari K.: Increasing intracellular cAMP and cGMP inhibits cadmiuminduced oxidative stress in rat submandibular saliva. Comp Biochem Physiol C Toxicol Pharmacol, 2003; 135C(3): 331-336. 187. Mc-Connell M. V., Aikawa M., Maier S. E., Ganz P., Libby P., and Lee R. T. : MRI of Rabbit Atherosclerosis in Response to Dietary Cholesterol Lowering . Arteriosclerosis, Thrombosis, and Vascular Biology, 1999; 19: 1956-1959. 188. Corti R., Osende J. I., Fallon J. T., et al.,: The selective peroxisomal proliferator-activated receptor-gamma agonist has an additive effect on plaque regression in combination with simvastatin in experimental atherosclerosis, in vivo study by high-resolution magnetic resonance imaging . J Am Coll Cardiol., 2004; 43: 464-473. 189. Erdoganl A., Luedders1 D. W., Muenz1 B. M., Schaefer1 C. A., Tillmanns1 H., Wiecha1 J., Ruediger C., and Kuhlmann W.: Sildenafil Inhibits the Proliferation of Cultured Human Endothelial Cells. International journal of Biomedical science, 2007; 3(2): pp.4. 190. Pasterkamp G., Kleijn D. P.V D., and Borst C.: Arterial remodeling in atherosclerosis, restenosis and after alteration of blood flow : potential mechanisms and clinical implications. Cardiovascular Research, 2000; 45: 843-852. 191. Langille B. L.: Arterial remodeling: relation to hemodynamics. Can J Physiol Pharmaco, 1996; l 74: 834-841. 192. Langille B. L., Bendeck M.P., and Keeley F.W.: Adaptations of carotid arteries of young and mature rabbits to reduced carotid blood flow. Am J Physiol, 1989; 256: 931–939. 193. Armstrong M.L., Heistad D.D., Marcus M.L., Megan M.B., and Piegors D.J.: Structural and hemodynamic responses of peripheral arteries of macaque monkeys to atherogenic diet. Arteriosclerosis, 1985; 5: 336-346. 194. Korshunov V. A., Schwartz S. M., and Berk B. C.: Vascular Remodeling: Hemodynamic and Biochemical Mechanisms Underlying Glagov’s Phenomenon, August 2007; Arteriosclerosis, Thrombosis, and Vascular Biology: Volume 27(8) pp 1722-1728. 195. Metelitsina T. I., Grunwald J. E., DuPont J. C., Ying G., and Liu C.: Effect of viagra on retinal vein diameter in AMD patients. Experimental Eye Research, 2006; 83: 128-132. 196. Yamashita A., Asada Y., Sugimura H., et al.,: Contribution of von Willebrand Factor to Thrombus Formation on Neointima of Rabbit Stenotic Iliac Artery Under High Blood-Flow Velocity. Arteriosclerosis, Thrombosis, and Vascular Biology, 2003; 23:1105. 197. Willens H. J.: Relationship of Peripheral Arterial Compliance and Standard Cardiovascular Risk Factors. Vascular and Endovascular Surgery, 2003; 37: 197-206. 198. Kocakoc E., Ardicoglu A., Bozgeyik Z., Kiris A., Yuzgec V., and Ogur E.: Effects of sildenafil on major arterial blood flow using duplex sonography. Journal of Clinical Ultrasound, 2005; 33(4): 173-175. 199. Ardicoglu A.: Hemodynamic effects of sildenafil citrate (Viagra) on segmental branches of bilateral renal arteries. Int. Urol Nephrol, 2005; 37(4):785-9. 200. García E. L., Ríos D. S., Martínez D. R., Dulín E., Fernández E. A., Fernández C. H. and López J. F. C.: Sildenafil as a Protecting Drug for Warm Ischemic Kidney Transplants: Experimental Results. THE JOURNAL OF UROLOGY, 2009;Vol. 182: 1222-1225. 201. Olin J. W., Piedmonte M., and Young J. R.: Duplex Scanning of Renal Arteries for Stenosis . Annals of internal medicine, 1996; 124 (3) : 370-371. 202. Schofield R. S., Edwards D. G., Schuler B. T., Estrada J., Aranda J. M., Pauly D. F., Hill J. A., Aggarwal R., and Nichols W. W.: Vascular Effects of Sildenafil in Hypertensive Cardiac Transplant Recipients. American Journal of Hypertension, 2003; 16:874–877. 203. Bude R. O., and Rubin J.M.: Relationship between the Resistive Index and Vascular Compliance and Resistance . Radiology, 1999; 211:411-417. 204. Samak G., Rao M. S., Kedlaya R. and Vasudevan D. M.: Hypolipidemic efficacy of ocimum sanctum in the prevention of atherogenesis in male albino rabbits. Pharmacology online, 2007; 2:115127. 205. Asgary S., Dinani N. J., Madani H., Mahzoni P., and Naderi G.: Effect of Glycyrrhiza glabra Extract on Aorta Wall Atherosclerotic Lesion in Hypercholesterolemic Rabbits. Pakistan Journal of Nutrition, 2007; 6:313-317. 206. Aversa A.: Strategies to Improve Endothelial Function and its Clinical Relevance to Erectile Dysfunction. european urology supplements, 2009; 8: 71–79. الخالصة: يعتبر مرض تصلب الشرايين القاتل الرئيسي على مستوى العالم .ويعتبر ارتفاع مستوى الدهون بالدم ,مرض السكري ,ارتفاع ضغط الدم الشرياني ,التدخين والسمنة من أهم عوامل الخطورة للمرض .أن) )sildenafil and vardenafilهي من األدوية األساسية التي تستخدم في عالج العجز الجنسي و من األدوية الواعده التي تستخدم في عالج تصلب الشرايين . الطريقة: اربعه وعشرون أرنبا محليا من الذكور أدخلت في هذه الدراسة .تم توزيع هذه األرانب بصورة عشوائية إلى أربعة مجاميع :المجموعة األولى هي مجموعة السيطرة الطبيعية وأعطيت غذاء قياسي طبيعي لمدة اثنا عشر أسبوعا .المجموعة الثانية أعطيت غذاء عالي الدسم يحتوي %2كولسترول لمدة اثنا عشر أسبوعا .المجموعة الثالثة أعطيت غذاء عالي الدسم لمدة ستة أسابيع ,بعد ذلك أعطيت عقار السلدنافيل 5ملم/كغم لكل يوم عن طريق الفم بالموازاة مع الغذاء العالي الدسم لمدة ستة أسابيع أخرى .المجموعة الرابعة أعطيت غذاء عالي الدسم لمدة ستة أسابيع ,بعد ذلك أعطيت عقار الفيردنافيل 18ملم/كغم لكل يوم عن طريق الفم بالموازاة مع الغذاء العالي الدسم لمدة ستة أسابيع أخرى .تم سحب عينات الدم أوال عند بداية الدراسة ,وعند ستة أسابيع من فترة الدراسة ثم كل أسبوع خالل فترة المعالجة البالغة ستة أسابيع .تم قياس مستوى الدهون بالدم وهي الكولسترول الكلي ,الكلسيريدات الثالثية ,الكولسترول واطئ الكثافة ,الكولسترول واطئ الكثافة جدا الكولسترول عالي الكثافة ,مقياس درجة التصلب ,ومستوى أالجهاد سمك جدار التاكسدي في الدم المتمثل بمستوى( )MDA and GSHتم قياس عرض و ُ الشريان األبهر البطني وتم قياس سرعة جريان الدم (السرعة االنقباضية العليا ,السرعة االنبساطية النهائية ,مقياس درجة المقاومة ,مقياس درجة االنقباضية واالنبساطية ونسبة السرعة االنقباضية العليا في الشريان الكلوي /السرعة االنقباضية العليا في الشريان األبهر) في الشريان األبهر ,الشريان الكلوي والشرايين الداخل كلوية بواسطة جهاز الدوبل رالمتوفر في العيادة الخاصة للدكتور عقيل زوين عند بداية الدراسة وبعد ستة اسابيع وعند نهايتها .كذلك تم فحص المقاطع النسيجية للشريان االبهر البطني عند نهاية الدراسة . وكانت النتائج كاألتي: سبب الغذاء العالي الدسم الذي يحتوي %2كولسترول زيادة معنوية ( )P<0.05في مستوى الكولسترول الكلي ,الكلسيريدات الثالثية ,الكولسترول واطئ الكثافة, الكولسترول واطئ الكثافة جدا ,الكولسترول عالي الكثافة ,وسبب زيادة معنوية ( )P<0.05في مقياس درجة التصلب .كما سبب زيادة معنوية ( )P<0.05في مستوى النخفاض أالجهاد التاكسدي في الدم المتمثل )بارتفاع مستوى MDAالمصاحب سمك الشريان االبهر و مستوى .) GSHكان هناك أيضا زيادة معنوية في عرض و ُ مقياس درجة مقاومتها ومقياس درجة االنقباضية واالنبساطية فيها كما سبب زيادة معنوية ( )P<0.05في السرعة االنقباضية العليا ,مقياس درجة المقاومة ومقياس درجة االنقباضية واالنبساطية في الشريان الكلوي والشرايين الداخل كلوية بالمقارنة مع مجموعة السيطرة الطبيعية .كما سبب زيادة معنوية ( )P<0.05في )نسبة السرعة االنقباضية العليا في الشريان الكلوي /السرعة االنقباضية العليا في الشريان األبهر( .لم يسبب الغذاء العالي الدسم زيادة معنوية ( )P>0.05في السرعة االنقباضية العليا والسرعة االنبساطية النهائية في الشريان األبهر كما لم يسبب زيادة معنوية ()P>0.05 في السرعة االنبساطية النهائية في الشريان الكلوي والشرايين الداخل كلوية. بالنسبة لنتائج الفحص النسيجي فان جميع األرانب التي تغذت على الغذاء العالي الدسم ظهر فيها مراحل مختلفة من تصلب الشرايين وكان هناك زيادة معنوية في نسبة التصلب ( )P<0.05بالمقارنة مع مجموعة السيطرة الطبيعية . لم يسبب العالج بالعقاران ) )sildenafil and vardenafilانخفاضا معنويا ( ) P<0.05في الكولسترول الكلي ,الكلسيريدات الثالثية ,الكولسترول واطئ الكثافة, الكولسترول العالي الكثافة ,الكولسترول واطئ الكثافة جدا في الدم ومقياس درجة التصلب .بينما سبب العقاران ))sildenafil and vardenafilزيادة معنوية ( )P<0.05في مستوى ( )GSHو انخفاضا معنويا في مستوى ( )MDAفي الدم. كما لم يسبب العالج بالعقاران ))sildenafil and vardenafilانخفاضا معنويا سمك جدار الشريان االبهر البطني.ولم يسبب العالج بالعقاران ( )P<0.05في عرض و ُ ))sildenafil and vardenafilانخفاضا معنويا ( )P>0.05في السرعة االنقباضية العليا ,السرعة االنبساطية النهائية ,مقياس درجة المقاومة ومقياس درجة االنقباضية واالنبساطية في الشريان األبهر ,ولم يسبب العالج بالعقاران sildenafil and ))vardenafilانخفاضا معنويا ( )P>0.05في) نسبة السرعة االنقباضية العليا في الشريان الكلوي /السرعة االنقباضية العليا في الشريان األبهر( ,بينما سبب العالج بالعقاران ) )sildenafil and vardenafilانخفاضا معنويا ( )P<0.05في السرعة االنقباضية العليا ,السرعة االنبساطية النهائية ,مقياس درجة المقاومة ومقياس درجة االنقباضية واالنبساطية في الشريان الكلوي والشرايين الداخل كلوية بالمقارنة مع مجموعة السيطرة الغير معالجة. بالنسبة لنتائج الفحص النسيجي العقاران ))sildenafil and vardenafilسببا انخفاضا في نسبة التصلب الشرياني ولكن هذا االنخفاض لم يكن انخفاضا معنويا ()P>0.05 بالمقارنة مع مجموعة السيطرة الغير معالجة. لم يكن هناك اختالف معنوي ( )P>0.05في الباراميترات المقاسة بين مجموعة األرانب التي عولجت بعقار sildenafilومجموعة األرانب التي عولجت بعقار . vardenafil االستنتاج : لم يسبب العقاران ) )sildenafil and vardenafilانخفاضا في مستوى الكولسترول الكلي ,الكلسيريدات الثالثية ,الكولسترول واطئ الكثافة ,الكولسترول واطئ الكثافة جدا ,الكولسترول العالي الكثافة في الدم ,كما لم يسببان انخفاضا في مقياس درجة سمك جدار الشرايين في األرانب المعالجة بهذين التصلب ,شدة تصلب الشرايين و ُ العقاريين ,بينما سبب العقاران ))sildenafil and vardenafilزيادة معنوية ( )P<0.05في مستوى ( )GSHو انخفاضا معنويا في مستوى ( )MDAفي الدم .لهذا فان هذين العقارين غيرا من قوة وصالبة الشرايين كما تبين من قدرتهما على تخفيض مقياس درجة المقاومة ومقياس درجة االنقباضية واالنبساطية في الشريان االبهر البطني,وكدلك تبين امكانية العقارين في تخفيض مقياس السرعة االنقباضية العليا, السرعة االنبساطية النهائية ,مقياس درجة المقاومة ومقياس درجة االنقباضية واالنبساطية في الشريان الكلوي والشرايين الداخل كلوية في األرانب المعالجة. دراسة س ُم ْك الطبقة الوسطية و الداخلية ثأثير السلدينافيل والفيردينافيل على ُ للشريان األبهر وعلى سرعة جريان الدم في الشريان األبهر ,الشريان الكلوي و الشريان الكلوي الداخلي في ذكور األرانب ذات مستوى الدهون المرتفع رسالة مقدمة إلى مجلس كلية الطب /جامعة الكوفة كجزء من متطلبات نيل درجة الماجستير في األدوية و العالجيات من قبل الصيدالني كرار حسين حريب كمونه بكالوريوس علوم صيدلة المشرف المشرف د .نجاح رايش الموسوي د.عقيل زوين أستاذ في علم األدوية والعالجيات مدرس في علم فسلجة القلب و األوعية الدموية 1430هــ 2009م