Large scale production of highly conductive reduced graphene
advertisement

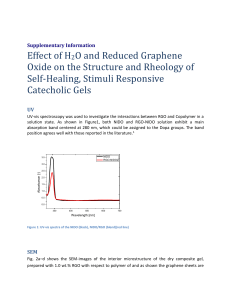
Supplementary Data Large-scale production of highly conductive reduced graphene oxide sheets by a solvent-free low temperature reduction Kyu Hyung Lee a, Byeongno Lee a, Son-Jong Hwang b, Jae-Ung Lee c, Hyeonsik Cheong c, Oh-Sun Kwon a, Kwanwoo Shin a, and Nam Hwi Hur a,* a b Department of Chemistry, Sogang University, Seoul 121-742, Korea Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 92115, USA c Department of Physics, Sogang University, Seoul 121-742, Korea * Corresponding author: Tel: +82-2-705-8440; Fax: +82-2-701-0967 E-mail address: nhhur@sogang.ac.kr (N. H. Hur). 1. General Information Materials. Graphite (<20 μm), potassium permanganate (KMnO4, ≥99.0%), heptane (CH3(CH2)5CH3, 99%), dodecane (CH3(CH2)10CH3, ≥99.0%), chloroform (CHCl3, ≥99.0%), dimethyl sulfoxide ((CH3)2SO, ≥99.9%), tetrahydrofuran (C4H8O, ≥99.0%), glycerol (HOCH2CH(OH)CH2OH, ≥99.9%) and hydrazine monohydrate (NH2NH2·H2O, 98%) were purchased from Sigma-Aldrich. Silicon oil was purchased from Shin-Etsu Chemical Co., Ltd. Concentrated sulfuric acid (H2SO4, assay 95%) and hydrogen peroxide (H2O2, assay 35%) was purchased from Jin Chemical Co., Ltd. All reagents were used without any further purification. Anodized aluminum oxide membranes (AAO, 200 nm pore size, 47 mm diameter) were purchased from Whatman. An inkjet printer was purchased from EPSON Korea Co., Ltd (EPSON Stylus T22). A4 size papers for inkjet printing were purchased from EPSON Korea Co., Ltd. (S042187) and Hyundai Printec Co., Ltd. (V2300). Solid hydrazine (H3N+NHCO2-) was prepared by the literature method [S1]. Briefly, solid hydrazine was obtained from the reaction of hydrazine hydrate with dry ice in an autoclave. Precaution. Solid hydrazine is very stable in a closed bottle. However, it could be harmful for health when it exposes in air. The equipment of effective ventilation is highly recommended for handling solid hydrazine to avoid vapor inhalation. Handle solid hydrazine inside a fume hood whenever it is grinding. Synthesis of graphene oxide (GO). Graphene oxide was prepared from graphite powder (Aldrich, <20um) by the modified Hummers method [S2,S3]. Briefly, graphite powder (5.0 g) and 130 mL of concentrated H2SO4 were added into a 1 L flask until the powder was completely dispersed. The flask was then cooled to 0 oC using a water-ice bath. A 15.0 g of KMnO4 powder was added to the cold reaction mixture, which was allowed to warm to room temperature. The temperature was then raised to 35 oC and the mixture was stirred for 2 h. The reaction mixture was cooled with an ice bath again, which was then diluted with 230 mL of water. To the diluted mixture, about 10 mL of H2O2 was added until evolution of gas was ceased. The mixture is allowed to settle for about 30 h. After settling, the clear supernatant was decanted. The remaining mixture was centrifuged and washed with a diluted HCl solution (10 % v/v) and a mixed solution containing CH3OH and water (50% v/v) several times. The resulting graphene oxide (GO) was dried under vacuum at room temperature for 24 h, yielding about 7.0 g of dark brown powders. Synthesis of reduced graphene oxide (RGO) by solid state reaction of GO with solid hydrazine. In a typical procedure, a 0.5 g of GO was mixed with 0.1 g of H3N+NHCO2-, which was then ground using a pestle and mortar. After grinding at ambient temperature, the ground powder was stored in a closed vessel. The ground mixture was allowed to react at 25 o C for 24 h or 50 oC for 10 min. Approximately 0.4 g of RGO was obtained from the vessel stored at 25 oC for 24 h while about 0.41 g of RGO was collected from the vessel stored at 50 o C for 10 min. Yields based on GO are 80 (25 oC) and 82 % (50 oC). Fabrication of freestanding RGO films. To prepare the GO films, the GO aqueous dispersion (3.5 g/L) was diluted with water to adjust the GO concentration (0.035 wt%). The GO films were prepared on a porous alumina membrane filter (200 nm pore size, 47 mm diameter; Whatman) with vacuum filtration method [S4]. Film thickness was tuned by the volume of the GO solution. Typically, about 10 mL of the GO solution was filtrated, followed by drying in the oven. The GO films were reduced by the vapor of solid hydrazine at 80 oC for 12 h in a closed bottle, which resulted in about 16 micron thick film. Preparation of GO dispersions for inkjet printing. To obtain uniform dispersions for inkjet printing, the GO dispersion (3.5 g/L) in water was mixed with glycerol. The optimal weight ratio of GO to glycerol is 5:4 (25 g of GO dispersion to a 20 g of glycerol). The mixed solution was sonicated for 1 h to obtain a very uniform GO dispersion suitable for inkjet printing. Ink-jet printing of GO dispersions. An EPSON Stylus T22 printer was used for dispensing the GO ink onto paper substrates. The printer employs the so-called drop-on-demand method in which the size and ejection of a drop is controlled by a piezoelectric transducer, compresses the fluid contained in a micro-capillary channel, jetting a submicron-sized (minimum size: 4 pL) drop from the orifice of the nozzle in a few microseconds [S5]. The designed images were ink-jet printed on A4 papers (EPSON, S042187) and PET films (Hyundai Printec Co., Ltd., V2300). Film thickness was controlled by printing repetitions. The printed films were placed in a closed bottle containing solid hydrazine, which were annealed at 80 oC for 12 h. Efficiency of solvent absorption by RGO. The efficiency of solvent absorption was evaluated by weight gain using a balance, where weight gain is defined as the weight of absorbed solvent per unit weight of RGO. Organic solvents were used for this experiment. For instance, a 0.53 g of RGO was immersed in heptane for 5 min. The weight of RGO absorbing heptane was 10.083 g. The weight gain for heptane was about 1,810%. Similar experiments were performed with dodecane, chloroform, silicon oil, dimethyl sulfoxide (DMSO), and tetrahydrofuran (THF). Their weight gains were also calculated and illustrated in Fig. S6. Instruments. Powder X-ray diffraction patterns were recorded with a Rigaku Miniflex diffractometer (Cu Kα) operating at 40 kV and 150 mA. Transmission electron microscope (TEM) was carried out on a JEOL JEM-2100F. Raman spectra of powder samples were obtained using a Jobin-Yvon Triax 550 spectrometer (1200 grooves/mm) equipped with a liquid-nitrogen-cooled charge-coupled-device (CCD) detector. A conventional confocal micro-Raman system was used. The 514.5 nm line of an Ar ion laser, with a total power of 1 mW, was focused onto the sample using a microscope objective (0.8 NA). The spectral resolution was ~1.2 cm−1. High resolution scanning electron microscope (HR-SEM) analyses were carried out using a Hitachi s-5500 microscope (Hitachi, Tokyo, Japan). X-ray photoelectron spectra (XPS) were carried out on an AXIS-NOVA (Kratos) with monochromatic Al Kα radiation. Thermo gravimetric analysis (TGA) was carried out using a TGA 2050 instrument. The sample was placed on a platinum pan for each run. The data were collected under N2 atmosphere from 25 ºC to 700 ºC at the rate of 5 ºC/min. Nicolet 205 instrument was used to measure infrared spectra. The elemental analysis was performed using a Vario Micro Cube, in which about 2.0 mg of each sample was subjected to 1,150 oC with surfanilic acid used as the standard. Adsorption and desorption measurements were carried out using an ASAP 2420 instrument (Micromeritics), with nitrogen as the adsorptive, at 77 K. The Brunauer-Emmett-Teller (BET) surface areas were calculated using p/p0 = 0.05-0.3 in the adsorption curve using the BET equation. The pore size distributions were obtained from the desorption curve using the density functional theory method. Prior to each sorption measurement, the sample was out-gassed at 300 oC for 24 h under a vacuum to remove all the impurities completely. 1H magic angle spinning (MAS) and 13 C MAS spectra with 1H decoupling were recorded at two different fields (4.7 T and 11.7 T) equipped with a Bruker DSX 200 console and a Bruker 7 mm CPMAS probe and a Bruker DSX 500 MHz console and a Bruker 4 mm CPMAS probe, respectively. All data acquisition was performed at room temperature. For single pulse experiments, a 4 microsecond 90 degree pulse was used for all nuclei. Sample spinning rates were 12-14 kHz at the 500 MHz spectrometer while 5-6 kHz was employed for 7 mm MAS probe. The chemical shifts were reported with respect to external references of tetramethylsilane for both 1H and 13 C nuclei. The conductivity was measured using a four-probe conductivity test meter (Keithely 2400) at room temperature. Measurements were carried out with a pressed pellet, a thick freestanding film, and an ink-jet printed paper with a rectangular shape. The topographic image was obtained using an AFM (NT-MDT NTEGRA Spectra). 2. Supplementary Figures Fig. S1 Time-dependent FT-IR spectra of mixed powders obtained from grinding GO and solid hydrazine powders. After grinding, the mixed powder was stored in a vial at room temperature without any agitation. Measurements were done after 12, 24, 36 and 48 h. In addition, FT-IR spectra are shown in the bottom and top panels corresponding to those of GO and graphite, respectively. They were included for comparison. Spike peaks, marked as asterisks, at approximately 2,300 cm-1 are due to CO2 in air. Fig. S2 FT-IR spectra of (a) graphite, (b) GO, and (c) reduced graphene oxide. Reduced graphene oxide was prepared by storing the ground powder of GO and solid hydrazine at 50 o C. FT-IR spectra of graphite and GO were included for comparison. Spike peaks, marked as asterisks, at approximately 2,300 cm-1 are due to CO2 in air. Fig. S3 C/O and C/N ratios of GO and RGO prepared by storing the ground powder of GO and solid hydrazine at 50 oC. Square symbols are obtained from elemental analysis data of GO and RGO. Fig. S4 TGA data of graphite (black line), GO (red line) and RGO (blue line) prepared by storing the ground powder of GO and solid hydrazine at 50 oC. For the measurements, temperature was increased by 5 oC per minute from 25 to 700 oC under N2 atmosphere. Fig. S5 Brunauer-Emmett-Teller N2 adsorption-desorption isotherms of RGO prepared by storing the ground powder of GO and solid hydrazine at 50 oC. Fig. S6 Cross-sectional SEM images of (a) GO layered film prepared by vacuum filtration of GO dispersion and (b) RGO film obtained from the reduction of the GO layered film by solid hydrazine at 80oC in a closed bottle. After reduction, volume was drastically expanded to produce foams between the layers. (c) A photograph showing that the resulting RGO film is easy to bend and is very flexible. In addition, the RGO film exhibits a shiny metallic luster. Fig. S7 Powder XRD patterns of (a) GO annealed at 50 oC in the absence of solid hydrazine and (b) RGO sheets obtained from the reduction of GO with solid hydrazine at 50 oC. 3. Supplementary Tables Table S1. Comparison of various solvent-based methods to produce RGO materials. Description Reagents Processable graphene sheets NH2NH2 (35 wt% in H2O) NH3 (28 wt% in H2O) Solvothermal reduction 1-methyl-2pyrrolidinone (NMP) Solvents useda) Ref. H2 O (~12,000 mL) [S6] refluxed at 205 C for 24 h annealed at 250 – 1000 oC H2 O (~6,000 mL) NMP (~6,000 mL) [S7] [S8] Reducing conditions heated at 95 oC for 1 h o Hydrazine reduction NH2NH2·H2O heated at 100 oC for 24 h H2 O (~7,500 mL) CH3OH (~4,500 mL) Chemical conversion NaBH4/H2SO4 heated at 80 oC for 1 h annealed at 1100 oC H2 O (>3,000 mL) [S9] Microwave exfoliation KOH (7 M in H2O) annealed at 800 oC for 1 h KOH (>150 mL) [S10] stored at 40 oC for 40 h CH3COOH (~1,125 mL) NaHCO3 (~375 mL), H2 O (~375 mL) (CH3)2CO (~150 mL) [S11] hydrothermally treated at 180 oC for 12 h annealed at 1050 oC for 3 h C4H5N (~375 mL) H2 O (~7,500 mL) [S12] Chemical graphitization Graphene framework HI CH3COOH C4H5N (pyrrole) a) Volumes of solvents were estimated assuming that 3.0 g of GO was used on the basis of corresponding procedures. 4. Supplementary References [S1] Lee B, Kang SH, Kang D, Lee KH, Cho J, Nam W, et al. Isolation and structural characterization of the elusive 1:1 adduct of hydrazine and carbon dioxide. Chem Commun 2011;47(40):11219-21. [S2] Hummers WS, Offeman RE. Preparation of graphitic oxide. J Am Chem Soc 1958;80(6):1339. [S3] Park S, An H, Piner RD, Jung I, Yang D, Velamakanni A, et al. Aqueous Suspension and Characterization of Chemically Modified Graphene Sheets. Chem Mater 2008;20(21):6592-4. [S4] Kong B-S, Yoo H-W, Jung H-T. Electrical conductivity of graphene films with a poly(allylamine hydrochloride) supporting layer. Langmuir 2009;25(18):11008-13. [S5] Kwon O-S, Kim H, Ko H, Lee J, Lee B, Jung C-H, et al. Fabrication and characterization of inkjet-printed carbon nanotube electrode patterns on paper. Carbon 2013;58:116-27. [S6] Li D, Müller MB, Gilje S, Kaner RB, Wallace GG. Processable aqueous dispersions of graphene nanosheets. Nat Nanotechnol 2008;3(2):101–5. [S7] Dubin S, Gilje S, Wang K, Tung VC, Cha K, Hall AS, et al. A one-step, solvothermal reduction method for producing reduced graphene oxide dispersions in organic solvents. ACS Nano 2010;4(7):3845-52 [S8] Stankovich S, Dikin DA, Piner RD, Kohlhaas KA, Kleinhammes A, Jia Y, et al. Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon 2007;45(7):1558–65. [S9] Gao W, Alemany LB, Ci L, Ajayan PM. New insights into the structure and reduction of graphite oxide. Nat Chem 2009;1(5):403–8. [S10] Zhu Y, Murali S, Stoller MD, Ganesh KJ, Cai W, Ferreira PJ, et al. Carbon-based supercapacitors produced by activation of graphene. Science 2011;332(6037):153741. [S11] Moon IK, Lee J, Ruoff RS, Lee H. Reduced graphene oxide by chemical graphitization. Nat Commun 2010;1:73. [S12] Zhao Y, Hu C, Hu Y, Cheng H, Shi G, Qu L. A versatile, ultralight, nitrogen-doped graphene framework. Angew Chem Int Ed 2012;51(45):11371-5.