Huntington`s Disease - University of Michigan
advertisement

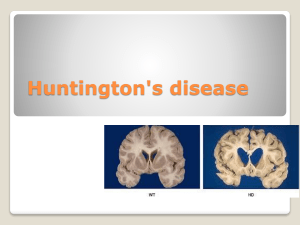
Huntington’s Disease Etiology/pathophysiology: -AD; due to mutation in huntingtin gene; based on anticipation of increasing CAG repeats. -Excess CAG repeats result in too much huntingtin proteins, causing mitochondrial toxicity, excitotoxicity, and immune activation. <26 repeats is normal, won’t get HD and won’t pass on HD -27-39 is equivocal, MAY get HD and MAY pass on HD >40 is inevitable huntington’s >50 CAG repeats tends to be juvenile onset -Prevalence 1/10000, but varies based on geographic location. -Natural history: Onset ranges from infancy to 80’s, average is 50’s; patients live 15-30 years on average from time of symptom onset. -Younger age of onset= faster the progression. -Medium spiny neurons most affected. -MRI with atrophy of caudate, though putamen is actually the main site involved. Symptoms: -Classic triad is movement disorder (usually chorea) + psychiatric disorder + dementia -Tend to present with chorea and personality changes first. -Chorea (motor impersistence, pendular reflexes, semi-purposeful movements, dance-like gait); improves as the parkinsonism worsens late in disease. Patients often unaware of debility. -Psychosis/dementia/depression/delusions/impulsivity/euphoria/OCD/suicide -parkinsonism (bradykinesia, rigidity) -Dysarthria/dysphagia -myoclonus -Tics -Dystonia (head, limbs, trunk) -Oculomotor apraxia (early finding; when patient saccades eyes, also thrusts head in that direction and blinks at same time) and square wave jerks -Ataxia -Anosognosia (underestimate or not acknowledge their deficits) Diagnosis: -Gene test; PRIOR to that, need to discuss consent, counseling, confidentiality, cost of test, and consequences (ie, HAVE PATIENT GET GENETIC COUNSELING PRIOR TO TESTING) -Can follow with the UHDRS (HD rating scale) -Consider investigating for mimics if gene test is negative or equivocal: Genetic DDx: -HDL1, HDL2, HDL3 •DRPLA •Neuroacanthocytosis •SCA 2, 3, 17 •NBIA •pantothenate kinase associated neurodegeneration (PKAN) -neuroferritinopathy aceruloplasminemia infantile neuroaxonal dystrophy •Benign hereditary chorea •Wilson disease •Mitochondrial disorders •Ataxia with oculomotor apraxia (types 1 and 2) •Ataxia-telangiectasia •Dystonia-ataxia-pyramidal (OPA3) Genetic workup for HD/mimics with chorea: Tx: 1) Education (disease, prognosis; see below): -Consider genetic counselors, particularly for other family members. Of note, for female HD patients wanting to have children, in vitro methods can create embryos that are guaranteed free of HD. 2) Symptomatic therapy: Psychiatric problems are often the most bothersome (chorea less so). -Close Psychiatric care and treatment is KEY if any hint of psychiatric manifestations. -SLP/PT/OT/nutrition if needed. -Anti-dopamine drugs for chorea (only use if bothersome): A) TETRABENAZINE: only FDA approved one (avoid if depressed or anxious, as can worsen these symptoms). B) Antipsychotics: Very reasonable first choice, particularly if concomitant psychiatric issues are present; be wary if h/o significant cardiac issues or prolonged QT. Olanzapine (start 5mg QHS, increase 7.5 10mg QHS if needed), Haldol, Risperdone. C) Amantadine (start 100mg daily, increase to 100mg TID). -Nothing works for neuroprotection or for dementia/cognitive symptoms (as of 2014) HD spiel that genetics clinic gives to patients: We explained that Huntington disease is a slowly progressive neurological condition that affects movement, coordination and thought processes. The symptoms of Huntington disease generally appear between 30 and 50 years of age though individuals can develop symptoms of Huntington disease at younger or older ages. Symptoms include involuntary movements, clumsiness, unsteady gait, deterioration in judgment and memory and in some individuals, personality changes, including depression and mood swings. The incidence of alcohol abuse and suicide is increased in individuals with Huntington disease. As Huntington disease progresses, involuntary movements, known as chorea, become more pronounced. Over time, speech and swallowing difficulties develop and the ability to think and remember events deteriorates. Once symptoms of Huntington disease have developed, generally an individual will survive 10-25+ years. Unfortunately, at the present time, there is no cure for Huntington disease. There are medications that can be taken to help with some of the symptoms but these medications cannot alter the course of Huntington disease. We also discussed the genetics of Huntington disease. We explained that our genetic information is contained in structures called chromosomes. Chromosomes are found in nearly all the cells in our bodies. Humans have 46 chromosomes which are organized into 23 pairs; one chromosome from each pair is inherited from each parent. Chromosomes are numbered 1-22, with the 23rd pair determining sex. Females have two X chromosomes; males have an X and a Y chromosome. Chromosomes contain thousands of genes. Genes are what determine physical traits, diseases and how our bodies function. The gene that is altered in individuals who have Huntington disease is located on chromosome number 4. Huntington disease is inherited in an autosomal dominant manner [autosomal-not related to gender so both males and females are affected; dominant-one altered copy of the gene required to have the condition]. Almost all individuals who have inherited the gene alteration for Huntington disease will develop Huntington disease. We explained that there is genetic testing available to determine if an individual has inherited the gene alteration for Huntington disease. The cost of the genetic testing is approximately $300 per sample and analysis can be performed on a blood sample. The Huntington disease gene contains a region of CAG repeats; individuals with Huntington disease usually have more than 39 copies of the CAG repeat on one of their two number 4 chromosomes though there is some variation between laboratories. Laboratories generally request that a blood sample from a family member with Huntington disease be analyzed first to confirm the presence of this CAG expansion prior to testing a family member at risk for Huntington disease. We emphasized that genetic testing for Huntington disease will not provide information on age of onset, type of symptoms, severity or disease progression. We also explained that in some cases genetic testing will not yield a definitive result. If the number of CAG repeats is in the inconclusive range, it will not be possible to predict whether or not an individual will develop Huntington disease. The Huntington Disease Society of America recommends that individuals at risk for Huntington disease have the following prior to testing: 1) genetic counseling 2) neurological evaluation and 3) psychological evaluation. Once these evaluations are completed and provided that there are no significant concerns, a blood sample can be drawn for genetic testing. If you decide to proceed with genetic testing, you will need to return to the Medical Genetics Clinic to have the blood sample drawn and to obtain your test results. Follow-up counseling sessions would be available. It is recommended that genetic testing be performed at a time of low stress when you are not experiencing major life changes. In addition, it is recommended that a "support person" such as a spouse, relative or a close friend be present when results are received and throughout the other evaluations if possible, and that a counselor in your community be identified for local support. The Huntington Disease Society of America has developed the guidelines for presymptomatic testing described above based on the experience of other at risk individuals for Huntington disease. We discussed the potential psychological implications of genetic testing for Huntington disease. Some individuals decide to be tested to make informed life decisions while others just feel a need to know their carrier status. Other individuals at risk for Huntington disease decide not to proceed with genetic testing because they do not want the burden of finding out that they will later develop a disease for which there currently is no cure. It is important that individuals take the time to carefully think through their decision about whether to proceed with genetic testing. It is not clear what the implications of being a carrier of Huntington disease could have on insurance coverage and employability. We discussed that it is important that you make sure that your health insurance, life insurance and disability insurance are up to date prior to undergoing genetic testing for Huntington disease. We also discussed the option of self-paying for the evaluations and testing. We explained that there will be a University of Michigan medical chart record of your visit to the Medical Genetics Clinic and discussed confidentiality issues. We provided you with information from the Huntington Disease Society of America. The phone number for the Huntington Disease Society of America is (800) 345-HDSA. There are Huntington disease support groups in Michigan that provide support for individuals with Huntington disease, their family members and caregivers. The phone numbers for the Michigan chapters are: Lansing(517) 321-6511; Mt. Clemens- (810) 465-7550. There are also Support Groups in: Kalamazoo-(616) 381-7725, Grand Rapids-(616) 949-9275 and Saginaw-(517) 835-9933. The Mid-Michigan HDSA chapter also sponsors a Help Line, which is coordinated by Janet Howes, MSW [(800) 909-0073], and has information about community resources. There is also a lot of information about Huntington disease on the Internet