ConfocalRaman_SuppInfo_Submission
advertisement

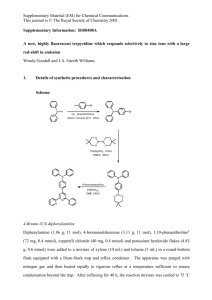
Optical Characterization and Confocal Fluorescence Imaging of Mechanochromic Acrylate Polymers M. Van Horn1, P. Smith1, B. P. Mason2, J. R. Hemmer3, J. Read de Alaniz3, J. P. Hooper1, S. Osswald1,#,* 1 Department of Physics, Naval Postgraduate School, 1 University Circle, Monterey, CA 93943 2 Research Department, Naval Surface Warfare Center, Indian Head Division, 3767 Strauss Ave., Indian Head, MD 20640 3 Department of Chemistry and Biochemistry, University of California, Santa Barbara, CA 93106 # Current Address: School of Materials Engineering, Purdue University, 701 West Stadium Avenue, West Lafayette, IN 47907 General. All reagents were obtained from Sigma Aldrich and Acros Organics and used without further purification. Proton NMR spectra were recorded on Varian spectrometers (at 500 or 600 MHz) and are reported relative to deuterated solvent signals. Gel permeation chromatography (GPC) was performed on a Waters Alliance HPLC System, with a 2690 Separation Module, detected by a Waters 2410 Differential Refractometer (RI). Elution was carried out on an Agilent, PLGEL 5μm, MIXED-D, 300x7.5 mm column in chloroform (0.25% triethylamine). Synthesis. Synthesis of 2,3,3-trimethyl-3H-indol-5-yl pivalate. Scheme S1. Preparation of 2,3,3-trimethyl-3H-indol-5-yl pivalate. 2,3,3-trimethyl-3H-indol-5-ol (1.00 g, 5.71 mmol) was placed in a flask and a septa was attached . The flask was flushed with nitrogen, then placed under a positive nitrogen atmosphere. Then dry dichloromethane (20 mL) was added, followed by triethylamine (854 μL, 6.85 mmol). Trimethyl acetyl chloride (843 μL, 6.85 mmol) was added dropwise. The reaction was allowed to run overnight. The reaction was quenched with 1N HCl, then extracted with dichloromethane. The aqueous layer was then neutralized with sodium bicarbonate and extracted with dichloromethane. The organic layers were then combined, dried over MgSO4, and the solvent removed under reduced pressure. Purified with flash column chromatography using a gradient of 10% to 40% ethyl acetate in hexanes (Yield 780 mg, 53%). 1H NMR (600 MHz, CDCl3) δ 7.50 (d, J = 8.2 Hz, 1H), 6.99 (d, J = 2.2 Hz, 1H), 6.97 (dd, J = 8.2, 2.3 Hz, 1H), 2.28 (s, 3H), 1.37 (s, 9H), 1.31 (s, 6H) ppm. Rf = 0.14 in 20% ethyl acetate in hexanes. Synthesis of 1,2,3,3-tetramethyl-5-(pivaloyloxy)-3H-indol-1-ium iodide. Scheme S2. Preparation of 1,2,3,3-tetramethyl-5-(pivaloyloxy)-3H-indol-1-ium iodide. 2,3,3-trimethyl-3H-indol-5-yl pivalate (780 mg, 3.01 mmol) and methyl iodide (5.6 mL) were combined and refluxed overnight. The reaction was allowed to cool, then was filtered and washed with benzene. The powder was collected and recrystallized from ethanol. The crystals were rinsed with diethyl ether (Yield 1.11 g, 93%). 1H NMR (600 MHz, DMSO-d6) δ 7.95 (d, J = 8.6 Hz, 1H), 7.68 (d, J = 2.3 Hz, 1H), 7.39 (dd, J = 8.6, 2.3 Hz, 1H), 3.96 (d, J = 1.2 Hz, 3H), 2.74 (d, J = 1.2 Hz, 3H), 1.52 (s, 6H), 1.33 (s, 9H) ppm. Synthesis of 8-hydroxy-1',3',3'-trimethyl-6-nitrospiro[chromene-2,2'-indolin]-5'-yl pivalate (monohydroxyl spiropyran) Scheme S3. Preparation of 8-hydroxy-1',3',3'-trimethyl-6-nitrospiro[chromene-2,2'-indolin]-5'-yl pivalate (monohydroxyl spiropyran) Tert-butyl acyl indolium iodide (300 mg, 0.75 mmol) and dihydroxy nitrobenzaldehyde (138 mg, 0.75 mmol) were combined in a round bottom flask and absolute ethanol (10 mL) was added. Then piperidine (150 μL, 1.50 mmol) was added and a reflux condenser was affixed to the flask. The reaction was then refluxed at 100 °C for an hour. The reaction was allowed to cool, and the solvent was removed under reduced pressure. Dichloromethane was added to the crude product, and this solution was washed with saturated aqueous ammonium chloride. The organic layer was dried with MgSO4 and the solvent was removed under reduced pressure. This crude product was used in the subsequent reaction without further purification. Scheme S4. Preparation of 8-((2-bromo-2-methylpropanoyl)oxy)-1',3',3'-trimethyl-6nitrospiro[chromene-2,2'-indolin]-5'-yl pivalate (Monofunctional Spiropyran) To crude monohydroxyl spiropyran 9 (approx. 100 mg, 0.228 mmol), 5 mL dry THF and pyridine (57 μL, 0.707 mmol) were added. Then α-Bromoisobutyryl bromide (85 µL, 0.684 mmol) was added and the reaction was allowed to run overnight. The reaction was then diluted with diethyl ether, filtered, and the solvent removed under reduced pressure. The crude material was purified by flash column chromatography using a gradient 2% to 10% ethyl acetate in hexanes. The pure fractions were then combined, and the solvent removed under reduced pressure. The residue was then dissolved in minimal dichloromethane and added dropwise to 10 mL of boiling hexanes. After leaving the hexanes solution in the freezer overnight, the product precipitated out as yellow crystals (Yield 60 mg, 45%). 1H NMR (500 MHz, Chloroform-d) δ 8.07 – 7.85 (m, 2H), 6.99 (dd, J = 10.5, 0.9 Hz, 1H), 6.79 (ddd, J = 8.3, 2.4, 0.9 Hz, 1H), 6.73 (d, J = 2.3 Hz, 1H), 6.46 (d, J = 8.3 Hz, 1H), 5.92 (d, J = 10.5 Hz, 1H), 2.66 (d, J = 0.9 Hz, 3H), 1.96 (d, J = 0.9 Hz, 8H), 1.58 (d, J = 8.3 Hz, 6H), 1.35 (d, J = 0.9 Hz, 9H), 1.25 (s, 3H), 1.20 (s, 3H). Synthesis of FF SP-PMA. Difunctional spiropyran synthesis and polymerization to form difunctional SP poly(methyl acrylate) (FF SP-PMA) was taken directly from the literature.1 Mn = 106 kDa, Mw/Mn = 1.31. Synthesis of MF SP-PMA. Polymerization to form monofunctional SP poly(methyl acrylate) (MF SP-PMA) was adapted from the literature1, substituting Monofunctional Spiropyran (above) for the fully functionized spiropyran derivative. Mn = 127 kDa, Mw/Mn = 1.33. 1. Potisek, S.L., et al., Mechanophore-linked addition polymers. Journal of the American Chemical Society, 2007. 129(45): p. 13808-13809.