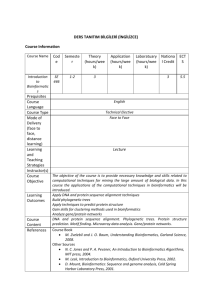
Sequence Searching & Alignments - European Bioinformatics Institute
advertisement

Bioinformatics Roadshow Sequence Searching and Alignments, May 2012 EBI Bioinformatics Roadshow Contents INTRODUCTION AND COURSE CONCEPT .......................................................................................................... 4 ABOUT THIS VERSION ...................................................................................................................................... 4 BIOINFORMATICS ............................................................................................................................................ 5 EMBL-EBI ......................................................................................................................................................... 7 NUCLEOTIDE SEQUENCES AND DATABASES .................................................................................................................... 8 ENA ................................................................................................................................................................ 8 PROTEIN SEQUENCES AND DATABASES ....................................................................................................................... 10 SEARCHING DATABASES ................................................................................................................................ 11 EBI SEARCH.......................................................................................................................................................... 12 EBI Search – advanced ................................................................................................................................. 15 DATABASE WEB PAGES ........................................................................................................................................... 17 HOMOLOGY VS. SIMILARITY..................................................................................................................................... 20 WHY MIGHT YOU NEED SEQUENCE SIMILARITY SEARCHING? ........................................................................................... 21 SEQUENCE SIMILARITY SEARCHING: AN OVERVIEW ....................................................................................................... 21 WAYS OF MATCHING SEQUENCES ............................................................................................................................. 22 Optimal Global Alignment ........................................................................................................................... 23 Optimal local alignment............................................................................................................................... 23 Substitution matrices ................................................................................................................................... 24 USING ALIGNMENT TOOLS AT THE EBI ........................................................................................................... 26 Step 1 – input ............................................................................................................................................... 27 Step 2 –parameters ...................................................................................................................................... 27 Step 3 – submit ............................................................................................................................................ 28 RESULTS .............................................................................................................................................................. 29 Results page ................................................................................................................................................. 29 Alignment ..................................................................................................................................................... 29 Submission details ........................................................................................................................................ 30 Implementing the Methods for Sequence Searching Tools: BLAST .............................................................. 33 Implementing the Methods for Sequence Searching Tools: FASTA .............................................................. 35 BLAST & FASTA Sensitivity ............................................................................................................................ 35 Sequence Searching Similarity tools at the EBI ............................................................................................ 37 USING FASTA ................................................................................................................................................. 39 Step 1 – database selection ......................................................................................................................... 40 Step 2 – input ............................................................................................................................................... 40 Step 3 –parameters ...................................................................................................................................... 40 Step 4 – submit ............................................................................................................................................ 41 RESULTS .............................................................................................................................................................. 42 Summary Table ............................................................................................................................................ 42 Tool Output .................................................................................................................................................. 43 Visual Output ............................................................................................................................................... 44 Functional Predictions .................................................................................................................................. 45 Submission Details ....................................................................................................................................... 45 Submit Another Job ...................................................................................................................................... 45 INTERPRETING AN ALIGNMENT ................................................................................................................................. 46 2 Sequence Searching and Alignments EBI Bioinformatics Roadshow USING BLAST.................................................................................................................................................. 49 NCBI BLAST parameters ............................................................................................................................... 49 WU-BLAST parameters................................................................................................................................. 50 BLAST RESULTS .................................................................................................................................................... 50 DIFFERENCES BETWEEN BLAST AND FASTA .............................................................................................................. 53 When to use what? ...................................................................................................................................... 53 PSI-BLAST....................................................................................................................................................... 54 PSI-BLAST Threshold ..................................................................................................................................... 55 FILTERS .......................................................................................................................................................... 58 HOMOLOGOUS OVER-EXTENSION (HOE) ....................................................................................................... 59 VECTOR CONTAMINATION ............................................................................................................................ 60 MULTIPLE SEQUENCE ALIGNMENT................................................................................................................. 62 CLUSTALW ..................................................................................................................................................... 63 RESULTS .............................................................................................................................................................. 66 Alignments ................................................................................................................................................... 66 Result Summary ........................................................................................................................................... 67 Jalview .......................................................................................................................................................... 67 Guide Tree .................................................................................................................................................... 68 Submission Details ....................................................................................................................................... 70 Submit Another Job ...................................................................................................................................... 70 Clustal Omega .............................................................................................................................................. 73 T-Coffee ........................................................................................................................................................ 73 MUSCLE ........................................................................................................................................................ 73 MAFFT .......................................................................................................................................................... 73 Kalign ........................................................................................................................................................... 73 WEBPRANK .................................................................................................................................................... 76 Results .......................................................................................................................................................... 77 RELATED ARTICLES FROM THE EBI ................................................................................................................. 79 FURTHER READING ........................................................................................................................................ 79 APPENDIX ...................................................................................................................................................... 80 NUCLEOTIDE CODES ............................................................................................................................................... 80 AMINO ACID CODES ............................................................................................................................................... 81 3 Sequence Searching and Alignments EBI Bioinformatics Roadshow Introduction and course concept Welcome to this Roadshow on Bioinformatics and more specifically, a series of resources and tools provided and/or hosted by the European Bioinformatics Institute (EBI) http://www.ebi.ac.uk/. This course is funded by the European Commission under SLING, “Serving Life-science Information for the Next Generation”. The EBI maintains the world’s most comprehensive range of molecular databases. As we move towards understanding biology at the systems level, access to large data sets of many different types has become crucial. Technologies such as genome-sequencing, microarrays, proteomics and structural genomics have provided ‘parts lists’ for many living organisms. This course in particular aims to provide you with an introduction to searching sequences and alignments at the EBI which we hope will be useful for you in your immediate or future research. There is really so much we would like to show you and at least make you aware that it exists, a few days, however, are not much, therefore do come and talk to us during the coffee and lunch breaks and of course, do not hesitate to ask questions. What we expect from YOU: In order to make sure you and your fellow participants can gain the most from this roadshow, do have a look at the signs below: HHHHHH!!! Switch off/silence your phones We hope you enjoy the course, and remember: The more you interact, the better the course will be! About this version v1.6 May 2012 Contributing authors Andrew Cowley – Bioinformatics trainer Hamish McWilliam – Software engineer Vicky Schneider – Training Programme project leader 4 Sequence Searching and Alignments EBI Bioinformatics Roadshow Bioinformatics Knowledge discovery starts by collecting, selecting and cleaning the data to finally fill a database. A database is a collection of files of consistent data that are stored in a uniform and efficient manner. A relational database consists of a set of tables, each storing records. A record is represented as a set of attributes which define a property of a record. Attributes can be identified by their name and store a value. All records in a table have the same number and type of attributes. Primary or archived databases contain information and annotation of DNA and protein sequences, DNA and protein structures and DNA and protein expression profiles straight from experimental results. Secondary or derived databases are so called because they contain the results of analysis on the primary resources themselves, including information on sequence patterns or motifs, variants and mutations and evolutionary relationships. Question 1: Which data resources do you know of? A very important characteristic of a database record is a unique identifier. This is crucial in Biology as in many other disciplines given the large amount of situations where an entity has many names, or a name can refer to multiple entities. The use of an accession number, a primary key derived by a reference database to describe the appearance of that entity in that database helps to overcome this problem. Unfortunately different databases may use their own conventions when assigning identifiers, so resolving which occur in difference resources can be a major challenge. The Protein Identifier Cross-Reference Service (PICR) at the EBI (http://www.ebi.ac.uk/Tools/picr/) is a tool to help find accession numbers and cross-references across databases. This is particularly useful when you are using programs to interface with these databases automatically. Database design is a critical step that covers: i) Defining (conceptual design) of the data requirements of the application, including the entities and their relationships. ii) Logical design is the implementation of the database using Data management systems (DBMS) which ensure the process to be scalable. iii) The physical design phase estimates the workload and refines the database design accordingly. During this phase design is optimized, indexing implemented and clustering approaches are optimized. These are fundamental steps in order to obtain fast responses to frequent queries without risking the database integrity. 5 Sequence Searching and Alignments EBI Bioinformatics Roadshow Top Challenges 1) Precise, predictive model of transcription initiation and termination: the ability to predict where and when transcription will occur in a genome (fundamental for HTS and Proteomics); 2) Precise, predictive model of RNA splicing/alternative splicing: ability to predict the splicing pattern of any primary transcript in any tissue (fundamental for Transcriptomics and Proteomics); 3) Precise, quantitative models of signal transduction pathways: ability to predict cellular responses to external stimuli (required in proteomics and pathways analysis); 4) Determining effective protein:DNA, protein:RNA and protein:protein recognition codes (important for recognition of interactions among the various types of molecules); Once we have the data we can then start examining it. For example, Clustering methods are used to identify patterns in the data, in other words to recognise what is similar, to identify what is different and from there to investigate whether differences are significant and biologically meaningful. Biological research tries to recognise patterns in order to infer relationships with previously-characterised sequences. It is essential to keep in mind that identifying similarity between sequences (e.g. nucleotide or amino acid sequences) is not necessarily equivalent to identifying other properties of such sequences, for example their function. A crucial step in bioinformatics is to pick the appropriate representation of the data. A simple but highly resourceful approach has been the use of controlled vocabularies (CVs), which provide a standardised dictionary of terms for representing and managing information. Ontologies are structurecontrolled vocabularies. As well as controlling the vocabularies, the data itself has to be standardised via different formats. Question 2: What data formats have you encountered before? 5) Accurate ab initio protein structure prediction (required for proteomics and pathways analysis); 6) Rational design of small molecule inhibitors of proteins (Chemigenomics); 7) Mechanistic understanding of protein evolution: understanding exactly how new protein functions evolve (comparative genomics); 8) Mechanistic understanding of speciation: molecular details of how speciation occurs (Comparative genome sequences, sequence variation); 9) Continued development of effective gene ontologies systematic ways to describe the functions of any gene or protein (genomics, transcriptomics, proteomics). With the advances in molecular biology, in particular the ever-growing progress in high throughput technologies, bioinformatics is continuously challenged on the ability to efficiently access large sets of data, allowing analysis of thousands items at a time, perhaps with complex constraints. Without bioinformatics it would be impossible to make sense of the huge amount of data produced (e.g. Omics research). Look at the increase in size of the EMBL Nucleotide Sequence Database (EMBL-Bank): Release 101 on 26-Aug-2009 contained 163,656,234 sequence entries comprising 283,748,816,763 nucleotides. Bioinformatics provides the structure that enabled storage of information in such a way that is retrievable, and comparable not only to similar data but also to other types of information. In 2002 Burge and colleagues produced a list defining in their view “Top 10 Future Challenges for Bioinformatics”. These currently do not essentially differ from those listed by Burge et al (2002), but simply stretched to meet the challenges inflict by the volume of data produced (see Panel on the left). 6 Sequence Searching and Alignments EBI Bioinformatics Roadshow EMBL-EBI The EMBL-EBI is one of the outstations of the European Molecular Biology Laboratory (EMBL). Today, the European Bioinformatics Institute (EBI) holds the world’s most comprehensive collection of databases and resources. EBI shares its central four mission objectives with EMBL, although focussed on bioinformatics rather than molecular biology: To provide freely available data and bioinformatics services to all facets of the scientific community in ways that promote scientific progress. To contribute to the advancement of biology through basic investigator-driven research in bioinformatics. To provide advanced bioinformatics training to scientists at all levels, from PhD students to independent investigators. To help disseminate cutting-edge technologies to industry. But how did it all start? Walgate (1982) communicated in Nature the pressure in Europe to set a suitable storing system for sequence data and the need to do it in collaboration to the relevant journals. One of the fundamental aspects of the discussion among interested parts was the establishments of a central data bank where sequences could be deposited and made freely available to the whole scientific community. Through “information engineering” the technical aspects were overcome, transforming rough drafts to the final computerised format we have today. 7 Sequence Searching and Alignments EBI Bioinformatics Roadshow Nucleotide sequences and databases The EMBL Nucleotide Sequence Database (also known as EMBL-Bank) constitutes Europe's primary nucleotide sequence resource. Main sources for DNA and RNA sequences are direct submissions from individual researchers, genome sequencing projects and patent applications. The database is produced in an international collaboration with GenBank (USA) and the DNA Database of Japan (DDBJ), forming the INSDC (International Nucleotide Sequence Database Collaboration). Each of the three groups collects a portion of the total sequence data reported worldwide, and all new and updated database entries are exchanged between the groups on a daily basis. The current database release (Release 103, March 2010), with accompanying release notes and user manual are available from the EBI servers. A publication in Nucleic Acids Research 2009 37: D19-D25 provides further information and details. The EMBL nucleotide sequence database, together with the Sequence Read Archive (SRA) and Trace Archive, forms part of the European Nucleotide Archive (ENA), and at the EBI it is maintained by the Protein and Nucleotide Database Group (PANDA) under Ewan Birney. ENA ENA captures and presents information relating to experimental workflows that are based around nucleotide sequencing. A typical workflow includes the isolation and preparation of material for sequencing, a run of a sequencing machine in which sequencing data are produced and a subsequent bioinformatic analysis pipeline. ENA records this information in a data model that covers: i) input information (sample, experimental setup, machine configuration), ii) output machine data (sequence traces, reads and quality scores) and iii) interpreted information (assembly, mapping, functional annotation). Data arrives at the ENA from a variety of sources. These include submissions of raw data, assembled sequences and annotation from small-scale sequencing efforts, data provision from the major European sequencing centres and routine and comprehensive exchange with our partners in the INSDC. Provision of nucleotide sequence data to ENA or its INSDC partners has become a central and mandatory step in the dissemination of research findings to the scientific community. ENA works with publishers of scientific literature and funding bodies to ensure compliance with these principles and to provide optimal submission systems and data access tools that work seamlessly with the published literature. Although the ENA has almost 30 years of history, the data and services are constantly changing to reflect growing volumes of data, ever improving sequencing technology and the broadening of applications to which sequencing is now put. 8 Sequence Searching and Alignments EBI Bioinformatics Roadshow As part of the global effort to improve access to and usability of nucleotide sequencing data, we collaborate extensively in the development of our services and technologies and in standards activities. Data submitted to ENA undergo a variety of validation steps that include automated quality checking and, where possible, manual inspection and curation. More information about the ENA and how to access the data can be found here: http://www.ebi.ac.uk/ena/ 9 Sequence Searching and Alignments EBI Bioinformatics Roadshow Protein sequences and databases Just as we have the ENA for nucleotide sequence information, the EBI also contains resources that deal with protein sequences. Most equivalent to EMBL-Bank is the UniProt Knowledge Base or UniProtKB. UniprotKB consists of two sections: TrEMBL is made up of automatically translated and annotated EMBL-Bank entries. Swiss-Prot is manually annotated, and importantly, reviewed, by experts. Because of this it’s much smaller than TrEMBL, but the entries are higher quality. As well as the core knowledgebase there are other supporting databases: UniRef contains sequences clustered together at different identity levels. UniParc is an archive of unique sequences (only), with a unique identifier for each one. Until a few years ago, EBI and SIB together produced Swiss-Prot and TrEMBL, while PIR produced the Protein Sequence Database (PIR-PSD). These two data sets coexisted with different protein sequence coverage and annotation priorities. TrEMBL (Translated EMBL Nucleotide Sequence Data Library) was originally created because sequence data was being generated at a pace that exceeded Swiss-Prot's ability to keep up. Meanwhile, PIR maintained the PIR-PSD and related databases, including iProClass, a database of protein sequences and curated families. In 2002 the three institutes decided to pool their resources and expertise and formed the UniProt Consortium. 10 Sequence Searching and Alignments EBI Bioinformatics Roadshow Searching databases Having the data in databases is one thing, but that’s only half the story - we need to be able to retrieve it for it to be useful. Question 3: What methods of searching databases do you know of? At the EBI there are many different ways of retrieving data. The easiest method is if you already have the unique identifier, as you can then perform a lookup on this information and retrieve the exact sequence of interest. But what if you don’t have that information? The information in sequence databases can be thought of as being contained in two parts – the sequence data itself, and the annotation or meta-data which talks about that sequence. This annotation could be anything from accession number and title to keywords describing something of interest about the sequence. If you are interested in a particular field then you can search for terms in a few different ways at the EBI. 11 Sequence Searching and Alignments EBI Bioinformatics Roadshow EBI search The EBI Search is on the main page, and the toolbar at the top of every page at the EBI, it allows you to very quickly search all the main databases in one go. Instruction: Open a browser and navigate to www.ebi.ac.uk It should look something like this: 12 Sequence Searching and Alignments EBI Bioinformatics Roadshow Instruction: Conduct a search for the term: BRCA1 The results will look something like this: The results are split into several categories, listed on the top left, while the main pane shows you example entries in each of the categories. They can be expanded by clicking the ‘View all results’ link 13 Sequence Searching and Alignments EBI Bioinformatics Roadshow at the bottom of the section. You can narrow results to a particular category by clicking on the category name in the top-left panel. You can then view each entry directly in the selected database by clicking on the name of the entry, or can use the View links to explore different ways of seeing the data, for example the raw text file, or through other databases at the EBI which look at the same data. Below the View links are the References – these are links to database entries that reference this sequence, for example an InterPro pattern or GO term. Instruction: Spend a minute exploring the results Question 4: How many entries are there in the nucleotide databases for BRCA1? 14 Sequence Searching and Alignments EBI Bioinformatics Roadshow EBI Search – advanced If you want more control over your search, you can perform an advanced search from the results page of any normal search by clicking the advanced search link. This brings up a control that allows you to search for exact phrases or to exclude specific words from the search. 15 Sequence Searching and Alignments EBI Bioinformatics Roadshow For even more control, you can search specific domains and fields by following the link from the advanced search page. 16 Sequence Searching and Alignments EBI Bioinformatics Roadshow If you really want to, you can form complex queries using the default search bar with the right query syntax. This is beyond the scope of this tutorial, but you can read more about it here: http://lucene.apache.org/java/2_4_0/queryparsersyntax.html Once you’ve done an advanced search the query syntax that made up the search is shown in the toolbar. Question 5: How many entries are there in the EMBL-Bank (last release, normal divisions) which have ‘BRCA1’ in their gene field? Why is this different from our answer to question 4? Database web pages So far we’ve looked at site-wide database searching, but each individual database (or group of similar databases) has its own web area at the EBI as well, and many of these contain their own powerful search methods. There are several routes to reach database web pages from the main EBI page. The most commonly used databases can be reached directly from the left hand column of links. 17 Sequence Searching and Alignments EBI Bioinformatics Roadshow The middle section contains links to groups of databases/resources organised by type. And of course there is the Databases drop down menu, which also contains links to groups of databases/resources organised by type: Finally if you’re looking for a specific database and can’t find it using the above, there is a full list of database web pages available by following the drop down from Databases > Databases Index and then selecting the Databases A-Z link on the left of the resulting page. 18 Sequence Searching and Alignments EBI Bioinformatics Roadshow Instruction: Spend a minute exploring different ways of navigating to databases 19 Sequence Searching and Alignments EBI Bioinformatics Roadshow Sequence searching & Alignments You will learn about: • Homology vs. Similarity • global and local score alignments. • substitution matrices (PAM & Blossum). • the algorithms used in search tools such as BLAST and FASTA. • Multiple sequence alignments (MSAs) • The tools available at the EBI for all of the above Homology vs. Similarity Since the mid-19th century, zoologists and botanists have learned to make a distinction between homologous organs (e.g. bat's wing and human's hand) and similar (analogous) organs (e.g. bat's wing and butterfly's wing). Homologous organs are not necessarily similar (at least the similarity may not be obvious); similar organs are not necessarily homologous. Phrases like “sequence (structural) homology”, “high homology”, “significant homology”, or even “35% homology” are as common, even in top scientific journals, and they are incorrect, considering the above definition. The term “homology” is used basically as a glorified substitute for “sequence (or structural) similarity”. 20 Sequence Searching and Alignments EBI Bioinformatics Roadshow Why might you need sequence similarity searching? Sequence similarity searching is used to infer relationships between sequences. Armed with a new piece of nucleic acid or protein sequence, you can compare it with all the sequences in the publicly available databases to find out whether anything similar or identical has been sequenced before. You can infer evolutionary relationships and functional information. Let’s imagine that you are studying the effects of rhinovirus infection on koalas. You have just sequenced a collection of expressed sequence tags (ESTs) from infected Rhinoviruses are the most koala bronchioles. There isn’t much koala DNA sequence in the public common viral infective domain, but nevertheless, you might expect to find that some of your EST agents in humans, and a sequences are identical to predicted transcripts from the genomic causative agent of the sequences of marsupials that have been sequenced. If these marsupial common cold. It is lytic in genes haven’t been very well characterised, nature. There are 99 ESTs are small portion of you could look for similarities between your recognized types of an entire gene that can be koala ESTs and those of a well-characterised rhinoviruses that differ used to help identify species, such as human or mouse. This might based on their varying unknown genes and to help you to identify some of the genes that surface proteins. map their positions within are expressed in your sample. You might also a genome. find some sequences that share only small regions of similarity to mammalian proteins, but bear a close resemblance to viral sequences. In short, you can learn a lot about the types of genes expressed in your sample through sequence similarity searching. The applications of sequence similarity searching are numerous, ranging from the characterization of newly sequenced genomes, through phylogenetics, to species identification in environmental samples. In this tutorial, we will guide you through the basic principles of how the most widely used sequence similarity programmes work, and will help you to practice using these tools. Sequence similarity searching: an overview All computational methods of sequence similarity searching try to align your query sequence with all of the sequences in a database. In the very simplest case, if we imagine our query sequence and a sample database sequence represented on two sides of a grid, we can plot which nucleotides (or amino acid residues) are identical in the two sequences. This is known as a dot plot. It’s immediately obvious that there’s more than one way of aligning even a short sequence. There are two different algorithms that are commonly used in creating dotplots: A Dot plot is a way of visualising a pairwise sequence alignment. A grid is created with a column for each position of one sequence and a row for each position in the other. Matches can then be marked in the appropriate square of the grid. 21 Sequence Searching and Alignments EBI Bioinformatics Roadshow 1) The first method involves matching identical regions of sequence and plotting a dot in these areas. 2) The second involves using “sliding windows” A score is calculated between two sequences using a to compare two sequences using a threshold two dimensional array of numbers called a matrix. score value. A window size is selected as a run The matrix for DNA is relatively simple, with each of adjacent nucleotide or amino acid residues, exact base match scoring 5 and each mismatch and a score chosen to reflect the degree of scoring –4. Various other scores represent matches similarity of sequence required. Each window between ambiguity codes. Matrices for amino acids of sequence A is compared to each window of are more complex, and are discussed later in the sequence B, and a dot is only placed in that chapter. region if the match scores or exceeds the set threshold level. Ways of matching sequences There is no unique, precise, or universally applicable notion of similarity. An alignment is an arrangement of two sequences, which shows where the two sequences are similar, and where they differ. An optimal alignment, of course, is one that exhibits the most similarities, and the least differences. Broadly, there are three categories of methods for sequence comparison. • Segment methods compare all overlapping segments of a predetermined length (e.g., 10 amino acids) from one sequence to all segments from the other. This is the approach used in dotplots. • Optimal global alignment methods allow the best overall score for the comparison of the two sequences to be obtained, including a consideration of gaps. These programs align sequences over their whole length. • Optimal local alignment algorithms seek to identify the best local similarities between two sequences also including explicit consideration of gaps. Alignment may only be over a short span of sequence. Question 6: Which of the following is a global alignment and which local? A B A T G T A T A C G C A - T G T A T A C G C A G T A T A - G C A G T A T A - - - G C 22 Sequence Searching and Alignments EBI Bioinformatics Roadshow Optimal Global Alignment Let’s look at one example for working out an optimal global alignment. Imagine we have the following query sequence and database sequence shown below. To work out the best alignment, we can assign a score to each dot in the grid, say 1 for every match, -3 for every mismatch and -5 for every gap. Adding these scores as we progress from top left to bottom right, and for each point choosing the maximum possible score, we produce a matrix with the maximum alignment score in the bottom right hand corner. Tracing back through the matrix from that maximal score reveals the alignment that produces the maximum score, this is the optimal global alignment. The NeedlemanWunsch algorithm, developed in 1970, works in this way. However, to do this we have to fill in the entire grid, which is computationally intensive and therefore slow. It also seldom works well for alignments across larger evolutionary distances because domains and motifs are shuffled. 1 2 3 4 5 23 Sequence Searching and Alignments EBI Bioinformatics Roadshow Optimal local alignment The earliest and most rigorous local alignment method is the Smith–Waterman method. This sets all negatively scoring cells in the grid to zero, making any positively scoring local alignments visible. The method then traces back from the highest scoring cell in the grid until it reaches a score of zero, the revealed local alignment is guaranteed to be the highest scoring local alignment between the sequences for the scoring scheme used. As with the Needleman-Wunsch method, the entire grid must be filled in, making this method very slow. Substitution matrices Sequence similarity searches of coding sequences have to take into account the fact that some mismatches are more conservative than others. For example, if one positively charged amino acid residue is substituted for another, protein function is less likely to be affected than if it’s changed to a hydrophobic residue. A conservative change should therefore incur less of a penalty than a radical change. A range of different substitution matrices has been developed for this purpose. Substitution of one amino acid for another with similar physicochemical properties is usually not selected against and represents a conservative change. Substitution of each amino acid for every other one can be given a score; conservative changes are not penalised heavily (e.g. substitution of Y for X scores –1 in this matrix) whereas a change to an amino acid with significantly different properties (e.g. F to P) is penalised more. The first of these, developed by Margaret Dayhoff and colleagues in 1978, is the Point Accepted Mutation (PAM) matrix. Based on the occurrence of observed amino acid replacements between closely related (>74% identity) protein sequences and normalising for divergence, Dayhoff et al. produced a matrix of expected substitution probabilities for proteins that are 1% diverged (the PAM1 matrix). Several versions of the PAM matrix have been derived by extrapolation from the 24 Sequence Searching and Alignments EBI Bioinformatics Roadshow PAM1 matrix, for example PAM40 (the expected substitution probabilities for 40% diverged proteins) and PAM250 (250% diverged). Because the PAM matrices were derived from alignments of closely related sequences, there are biases favouring the substitutions of amino acids that can be achieved through single nucleotide changes in the genetic (codon) code. These biases are not relevant over larger distances because there is plenty of time for multiple nucleotide changes, so PAM matrices are more appropriate for searching or aligning more closely related sequences. BLOSUM (Blocks of Amino Acid Substitution Matrix) developed by Henikoff and Henikoff in 1992, uses frequency tables of substitutions observed in multiple alignments. BLOSUM50 scores according to an alignment of proteins with 50% overall identity; BLOSUM62 uses an alignment of proteins with 62% identity. In contrast to the PAM matrices, larger BLOSUM numbers therefore represent smaller evolutionary distances. 25 Sequence Searching and Alignments EBI Bioinformatics Roadshow Using alignment tools at the EBI Let’s have a go using these algorithms for ourselves. Instruction: Navigate to the EBI pairwise sequence alignment page You can either type in an address directly (www.ebi.ac.uk/Tools/psa/) or use the Tools drop down menu. You will be faced with a choice of programs, split into global or local alignments. 26 Sequence Searching and Alignments EBI Bioinformatics Roadshow Once you chose a program, the input page looks like this: Input for pairwise alignments is very simple Step 1 – input Here you can enter your two sequences. Most common formats are recognised, but please don’t try to invent your own using Word! You can either paste/type the sequence directly, or upload a file containing it from your computer using the browse button. Step 2 –parameters The program will be set up with some default parameters, however you can change them if you wish. You can click on any parameter title to get help on it. The main options are: 27 Sequence Searching and Alignments EBI Bioinformatics Roadshow Matrix The comparison matrix to be used to score alignments when searching the database Gap open Penalty to start a gap in the alignment Gap extend Penalty for each base or residue in the gap Step 3 – submit When you’re happy with everything else, select submit to run the job. 28 Sequence Searching and Alignments EBI Bioinformatics Roadshow Results Results page The results page is fairly simple. At the top of the page are some tabs that switch between the alignment, submission details, and the form to submit a new job. parameters used for the job are shown, and there is a link to the output file. The output file text is also displayed further down the page. Alignment This page shows the results of the alignment. The table gives a summary of the results: Length reports the length of alignment. Identity reports the number of identical residues that are found aligned between the two sequences. Similarity reports the number of aligned residues that score positively in the substitution matrix (ie similar types of residues). Gaps reports the number of gaps inserted into the alignment. 29 Sequence Searching and Alignments EBI Bioinformatics Roadshow Score gives the literal score of the alignment as worked out by the algorithm. Then the alignment itself is displayed: The top line is the first sequence and the bottom is the second sequence. Gaps in sequence are displayed with a ‘–‘ character. Where two identical residues line up they are connected with a ‘|’ where two very similar residues line up they are connected with a ‘:’. Where less well conserved substitutions are made they are connected with a ‘.’. Submission details This page gives details of how the program was run. It tells you what version of the tool was run, when it was launched, details of your input and the tool output, as well as the original command line used to launch the job and details of the selected parameters. These can all be useful if you need to recreate a job on your local machine, or to repeat an alignment in the future. This page is also very useful to us if you have a problem and need to contact us. Instruction: Try running your own global alignment using the provided sequences: Use the EMBOSS needle program Leave the parameters set at their defaults 30 Sequence Searching and Alignments EBI Bioinformatics Roadshow Question 7 (global): What is the Length, Score, %identity, %similarity and %gaps of the alignment? Length Score Identity% Similarity% Gaps% Now let’s try a local alignment. Instruction: Try running your own local alignment using the provided sequences: This time use the EMBOSS water program Leave the parameters set at their defaults Question 8 (local): What is the Length, Score, %identity, %similarity and %gaps of the alignment? Length Score Identity% Similarity% Gaps% 31 Sequence Searching and Alignments EBI Bioinformatics Roadshow Question 9: In words, how would you describe the key differences between the global and local alignment results? Can you think of ways to improve the alignment? Now let’s try changing the matrix parameter. Instruction: Try running your own local alignment using the provided sequences: Use the EMBOSS water program Change the matrix to BLOSUM 40, and leave the other parameters at their defaults Question 10: What is the Length, Score, %identity, %similarity and %gaps of our new alignment? Can you describe what has happened? Length Score Identity% Similarity% Gaps% 32 Sequence Searching and Alignments EBI Bioinformatics Roadshow Alignment against databases While it’s possible (and very accurate) to run optimal alignments against databases (using SSearch or GGsearch at the EBI for example), the computational requirements are such that it takes a very long time, and uses a large amount of memory. It is more practical to use a heuristics based method such as BLAST or FASTA. Implementing the Methods for Sequence Searching Tools: BLAST BLAST, which stands for Basic local alignment tool and was developed by Altschul and colleagues in 1990. BLAST uses an approximation of the Smith–Waterman algorithm which makes is quite fast, however this gain in speed is offset by a decrease in accuracy. Unlike the true Smith–Waterman algorithm, BLAST is not guaranteed to find the optimal alignment between your query sequence and the test sequences. However, it will find good alignments and provides a statistical means of gauging your confidence in each alignment: (1) It searches for ‘words’ of a user-defined length (the shorter the word, the more sensitive the search). (2) It then extends these words in both directions until it finds a mismatch. (3) It then performs an approximation of the Smith–Waterman algorithm to create a gapped alignment between the query sequence and the test sequence. (4) Finally it calculates and reports the probability of the alignment occurring by chance. 33 Sequence Searching and Alignments EBI Bioinformatics Roadshow When doing a Blast search one can set the “expectation threshold” (EXP THR) which establishes a statistical significance threshold for reporting database sequence matches. EXP THR is interpreted as the upper limit on the expected frequency of a chance occurrence of a match within the context of the entire database search; in other words, it sets an upper limit on the E-value. Any database sequence whose BLAST alignment to the query sequence satisfies EXP THR is reported in the output file. An alignment with an E value of ≥1.0 is expected to be found at least once by chance in the searched database and an E value of ≥5.0 is expected to be found at least five times (see figure below). Raising this threshold increases the likelihood of reporting distantly related matches, but the frequency of chance matches reported will tend to grow at a much faster rate than real matches with EXP THR set above 1.0. 34 Sequence Searching and Alignments EBI Bioinformatics Roadshow Implementing the Methods for Sequence Searching Tools: FASTA David Lipman and William Pearson (1988) developed FASTA which gets around the speed problem by: (1) FASTA breaks the query and test sequences into overlapping words and looks for exact matches. (2) Then it re-scores these matches using a substitution matrix. (3) Next it tries to join the highest-scoring segments. A ‘joining threshold’ set by the user eliminates segments that are unlikely to be part of the alignment. (4) Finally, FASTA uses the Smith–Waterman method to optimise the alignment, using only the part of the matrix that contains the top-scoring segments. BLAST & FASTA Sensitivity 35 Sequence Searching and Alignments EBI Bioinformatics Roadshow FASTA therefore provides a means of performing a sensitive search against a large database in a reasonable time. Nowadays, it is possible to approach the sensitivity of a FASTA search by using BLAST and setting high sensitivity values. However, the Sensitivity: There is a trade-off between alignment statistics for FASTA can be considered more sensitivity and search speed. Increasing the robust than those for gapped-BLAST. This is because sensitivity makes the search more FASTA produces and scores an alignment of the query computationally intensive and therefore slower. sequence with a large sampling of the database, giving it Decreasing the sensitivity, for example when a distribution of scores that represents the entire you are looking for almost exact matches, can database:sequence range of alignments. BLAST is fast dramatically increase the search speed. There is because it does not bother producing an alignment for also a trade-off between sensitivity and most database sequences; alignments are only specificity: increasing sensitivity tends to triggered if the initial word-match criteria are met. decrease specificity (greater propensity for Consequently, BLAST does not have a complete chance matches). So for example: if you are distribution of alignment scores over the database from looking for vector contamination you will which to calculate the significance of the reported choose low sensitivity, whereas if you are matches. Instead, BLAST uses pre-computed values for looking for long distance related sequence you will opt for high sensitivity. the score distribution rather than calculating values that are specific to the search carried out. 36 Sequence Searching and Alignments EBI Bioinformatics Roadshow Sequence Searching Similarity tools at the EBI The EBI provides you the option to use several sequence searching similarity tools at www.ebi.ac.uk/Tools/sss. These are maintained by the External Service (ES) team. The ES team puts considerable effort in making sure to provide you with the state of the art tools in sequence searching and also with the flexibility to tailor your queries to the most appropriate search parameters. Therefore you will find several options not only for the types of tools but also within the tools, the amount of variable and parameters you can change. 37 Sequence Searching and Alignments EBI Bioinformatics Roadshow We have talked about BLAST and FASTA but what are all the variations above? A quick way to make a distinction among these tools is to label them by being either heuristic or rigorous. A fundamental challenge in computer science is to make algorithms that find verifiable good solutions using a proved bounded amount of computation time. A heuristic algorithm gives up one or both of these goals. In other words heuristic is an algorithm that is able to produce an acceptable solution to a problem in many practical scenarios, but for which there is no formal proof. Heuristics are typically used when there is no known method to find an optimal solution, under the given constraints (of time, space etc.) or at all. Rigorous on the other hands means to applying an algorithm that can produce proof, that gives you the most optimal solution, therefore it is exhaustive and should provide you with the best answer, however it is slow. Following these definitions, the Sequence Searching Tools mentioned above are label as shown on this panel: 38 Sequence Searching and Alignments EBI Bioinformatics Roadshow Using FASTA Let’s have a go using FASTA for ourselves. Instruction: Navigate to the EBI sequence search tools page You can either type in an address directly (www.ebi.ac.uk/Tools/sss/) or use the Tools drop down menu. At the time of writing the tools drop down points to our old The framework from framework while the address goes to the new one. You can click the link to which we launch our tools was revamped access the new one, which we will be using in this tutorial. FASTA is launched by picking a type of database listed next to the FASTA tool – this sets up the defaults appropriately for your choice, but you can always change the database once in the tool if you wish. Instruction: recently, and now forms a common basis for many programs. For more information about this framework see: A new bioinformatics analysis tools framework at EMBL–EBI (2010) Goujon et al. doi:10.1093/nar/gkq313 Select the Protein database for FASTA You should end up at a screen that looks like the following: 39 Sequence Searching and Alignments EBI Bioinformatics Roadshow This screen will hopefully look quite familiar to you when conducting other searches as well. There are four steps to submitting a job. Step 1 – database selection Here is where you select which databases to search against. You can select more than one, and you can also expand subsections to narrow your search by taxonomic division for example. If you changed your mind and want to do a different type of search (for example, against nucleotide sequences) then you can select that here as well and it will reset the form. Step 2 – input Here you can enter your query sequence. Most common formats are recognised, but please don’t try to invent your own using Word! You can either paste/type the sequence directly, or upload a file containing it from your computer using the browse button. Step 3 –parameters Most important here is choice of program – FASTA is grouped together with others like SSEARCH (because they were authored by the same person and come in the same package). It should be set up correctly according to the button you pressed to get to this page. The program will be set up with 40 Sequence Searching and Alignments EBI Bioinformatics Roadshow some default parameters, however to look at these or change them you need to click the ‘More options’ button. You can click on any parameter title to get help on it. Here is a summary: Matrix The comparison matrix to be used to score alignments when searching the database Gap open Penalty to start a gap in the alignment Gap extend Penalty for each base or residue in the gap Ktup ‘Word size’ used to identify runs in the first stage of alignment Expectation upper value This allows you to ignore results that have above a certain expectation score (ie become more distant) Expectation lower value This allows you to ignore results below a certain expectation (ie ignore close relatives) DNA strand When searching DNA you can specify which strand is used. By default both are searched Histogram Turn on/off the display of statistical histogram in FASTA results Filter Which low complexity filter to use Statistical estimates Which statistical method to use to evaluate values used in the Expect score calculation Scores Maximum number of scores reported in the summary Alignments Maximum numbers of alignments reported in the summary Sequence range Allows you to specify which portion of the query sequence to use in the search Database range Allows you to cut back on database sequences searched against by specifying a number of residues range Step 4 – submit When you’re happy with everything else, here is where you click to submit the job. For longer runs it is recommended that you tick the box to send you an email when the job is complete. This will contain a link back to the results so there is no need to keep your browser open. Email jobs are usually stored on the servers for longer as well, while interactive job results are deleted more quickly. 41 Sequence Searching and Alignments EBI Bioinformatics Roadshow Once you’ve submitted your job the first thing that happens is that the input is validated – few things are more frustrating than preparing a job and firing it off to then check your email later in the day and find that you made a minor mistake somewhere and the job failed. If everything is okay then the job will run and you will see a job running screen if you ran it interactively. Eventually the job will finish and you will either be taken to the results page automatically or emailed a link to it. Results Summary Table There is a lot of information to take in for the results page! You are first presented with the summary table which quickly lists the top results in a table format. You can change the ordering of the table by clicking the arrows next to each column header – by default they are ranked by Expect value or E(). The first column contains a tick box which allows you to select database results for further actions, for example to view the sequence annotation or detailed alignment with the query sequence. You can also use the buttons to clear selections, select all, or invert selection. To download the selected sequences click the download button. The second column (DB:ID) gives the database ID of the sequence. The third column (Source) gives some quick information about the sequence, as well as cross references that have been found referring to it in other resources across the EBI – so you can quickly look up more information about the sequence in these resources. 42 Sequence Searching and Alignments EBI Bioinformatics Roadshow Length reports the length of the database sequence. Score gives the literal score of the alignment. Identities reports the number of identical residues that are found aligned between the query and database sequence. Positives reports the number of aligned residues that score positively in the substitution matrix (ie similar types of residues). E() gives the Expect score for the alignment – this is a measure of how likely you are to find that alignment by chance. When the numbers are very small it reports them as 1.0E-10 for example. This is the same as 1e-10, or 0.0000000001. Tool Output This tab switches the view to the raw, original output from FASTA – this can be useful when you want to view the full text output from the program in case it contains something the summary or other pages don’t cover. You can download it as a text or XML file. Clicking on the icon will jump you straight to the alignment. Clicking on the sequence ID will take you to the original database entry for that sequence. 43 Sequence Searching and Alignments EBI Bioinformatics Roadshow Visual Output This output gives you a nice way of visualising which portions of the sequence are aligning, as well as colour-coding the alignment by Expect score. Hovering your mouse over the sequence ID on the left-hand side will show a guide box around the alignment. Clicking the mouse will take you to the alignment (from the raw output of the program). 44 Sequence Searching and Alignments EBI Bioinformatics Roadshow Functional Predictions This tab is another example of how we are bringing together different resources at the EBI to give you extra information. It searches a variety of resources for family and domain predictions and shows you the results graphically, so you can easily see which portions of your sequence and alignments correspond to these features. Again, you can click on the links on the left-hand side to jump to the entry information at the resource. Submission Details This page contains all the original parameters used to launch your job, together with easy links to the exact input used and the output results. This information is really useful for several reasons: If you want to repeat a job then you might want to use exactly the same parameters; if you’re interested in running the command line version of the tool then this will give you the exact command line used; and finally if you need help or support then this page contains all of the information you need to give us to be able to help you quickly. Submit Another Job The final tab just quickly takes you back to the page where you can start a new FASTA job. 45 Sequence Searching and Alignments EBI Bioinformatics Roadshow Interpreting an alignment The figure below shows a typical alignment Key - Gap : Identity . Similarity X Filtered The header shows some information about the database sequence, followed by some of the raw scores from the program itself. The key bits of information are the E() score, the % identity and % similarity numbers. Below that is the actual alignment itself – the top line is the query sequence and the bottom is the database sequence. Gaps in sequence are displayed with a ‘–‘ character (none in the above alignment). Where two identical residues line up they are connected with a ‘:’ where two similar residues line up they are connected with a ‘.’ Instruction: Try running your own FASTA search using the provided sequence: Search against the UniProtKB/Swiss-Prot database only – if you search against the full UniProt Knowledgebase it will take a very long time! Make sure the FASTA program is selected Leave the parameters set at their defaults 46 Sequence Searching and Alignments EBI Bioinformatics Roadshow Question 11: What are the default gap open, gap extend, ktup and matrix parameter settings for this search? Question 12: What can you say about the likely function of this protein? Question 13: What are the DB:IDs, Scores, %identities, %positives and E() of the top two results? ID Score Identity% Positives% E() 47 Sequence Searching and Alignments EBI Bioinformatics Roadshow Question 14: Have a look at the actual alignments with the top two results – what can you say about them? 48 Sequence Searching and Alignments EBI Bioinformatics Roadshow Using BLAST Now that we are familiar with running FASTA searching, using BLAST should be very easy – the interface is effectively the same. Simply select a database next to the BLAST tool of interest to enter the tool. NCBI BLAST This version of BLAST is the version maintained at the NCBI WU-BLAST This version of BLAST was created by Dr Warren Gish, formerly of Washington University Both versions can trace their history back to the same algorithm, but were developed separately, often implementing ideas that first appeared in the other version. The parameters are handled slightly differently in each case: NCBI BLAST provides access to parameters like those used by FASTA or SSEARCH (eg gap open, gap extend). WU-BLAST hides direct access to those, but instead provides a sensitivity parameter which combines several adjustments at different stages of the algorithm. The raw results are slightly different between the two, but as we parse the results they will appear the same to you (unless you look at the Tool Output page). NCBI BLAST parameters Matrix The comparison matrix to be used to score alignments when searching the database Gap open Penalty to start a gap in the alignment Gap extend Penalty for each base or residue in the gap Exp. Thr (expectation threshold) This allows you to ignore results that have above a certain expectation score (ie become more distant) Filter Which low complexity filter to use Drop off Controls how far a potential HSP is allowed to extend Scores Maximum number of scores reported in the summary Alignments Maximum numbers of alignments reported in the summary Sequence range Allows you to specify which portion of the query sequence to use in the search Gap align Allows gapped extensions of alignments Alignment views Options for formatting the alignment output (Tool Output) 49 Sequence Searching and Alignments EBI Bioinformatics Roadshow WU-BLAST parameters Matrix The comparison matrix to be used to score alignments when searching the database Exp. Thr (expectation threshold) This allows you to ignore results that have above a certain expectation score (ie become more distant) Filter Which low complexity filter to use View filter Display any sequence filtered out (in Tool Output) Sensitivity General parameter affecting search sensitivity – this makes adjustments to several internal parameters Scores Maximum number of scores reported in the summary Alignments Maximum numbers of alignments reported in the summary Sort Choose which value to sort the Tool Output results by. Stats Choice of statistic methods used in generation of Expect statistics topcomboN In WU-BLAST HSPs are classified into a number of sets, you can use this parameter to restrict the display to only the N highest scoring sets. Alignment views Options for formatting the alignment output (Tool Output) BLAST results Unsurprisingly, the results from BLAST runs on our servers are displayed in the same way as FASTA results, and all the tabs are equivalent. The main differences in format will only appear if you look at the raw output via the Tool Output page. 50 Sequence Searching and Alignments EBI Bioinformatics Roadshow Key - Gap [residue] Identity + Similarity X Filtered The above picture shows the alignment section of an NCBI BLAST run. This time identical residues are actually listed between the two sequences, which are labelled Query for query sequence and Sbjct (subject) for database sequence. Similar residues (Positives) are indicated with a +. The number of gaps inserted into the overlapping sequence regions is also reported. Instruction: Try running your own NCBI BLAST search using the provided sequence: Search against the UniProtKB/Swiss-Prot database only – if you search against the full UniProt Knowledgebase it will take a very long time! Make sure the BLASTP program is selected Leave the parameters set at their defaults 51 Sequence Searching and Alignments EBI Bioinformatics Roadshow Question 15: What are the default gap open, gap extend, drop off and matrix parameter settings for this search? Question 16: What are the DB:IDs, Scores, %identities, %positives and E() of the top two results? ID Score Identity% Positives% E() Question 17: Are these different from our FASTA search earlier? 52 Sequence Searching and Alignments EBI Bioinformatics Roadshow Differences between BLAST and FASTA The following table summarises the key differences between BLAST and FASTA BLAST FASTA Fast Good with proteins Might miss potential alignment Not as fast as BLAST Good with proteins and DNA Aligns against all database sequences Produces S&W alignments Good for cousins Produces HSPs Good for siblings When to use what? In general the larger the database the faster the algorithm you should use, and likewise the larger the query sequence the faster the algorithm you should use. For very small queries or databases then dynamic programming methods like SSEARCH can be great. 53 Sequence Searching and Alignments EBI Bioinformatics Roadshow PSI-BLAST Position-Specific Iterative BLAST, or PSI-BLAST, is a clever tool which allows you to create your own custom scoring matrix based on the conservation of residues you find in your own searches, rather than some model made with different sequences. PSI-BLAST workflow It starts with a normal BLAST, however you can then 1. Normal BLAST search select which sequences in the results will be used to build a profile. These sequences are then aligned and conserved residues at each position are scored more highly in a new type of matrix which allows for different scores at different positions in the sequence. A new BLAST search is run with this matrix, called a Position Specific Scoring Matrix or PSSM. The results can themselves be used to create another PSSM for another run, and so the process is iterative. The aim of PSI-BLAST is concentrate the alignment on positions that are important, while allowing for more variability in areas that aren’t so important. So a functional area or binding motive might be more important than sequence that forms part of a loop for example. 2. Align selected results Searches made with a PSSM can find matches with sequences that were scored too low to be considered in a normal BLAST search, but have scored more highly with the new matrix – these are marked as ‘new’ by the PSI-BLAST tool. 3. Create PSSM You can also save your search and continue it at a later date, or save the PSSM itself. (Continued on next page) 54 Sequence Searching and Alignments EBI Bioinformatics Roadshow PSI-BLAST workflow (cont.) The parameters for PSI-BLAST are the same as NCBI BLAST, with the addition of a new threshold: 4. Use PSSM to score new BLAST alignment PSI-BLAST Threshold This expectation value controls the default selection of sequences to be used for generation of the PSSM – sequences scoring higher than this (ie don’t align as well) won’t be included. Once the first iteration is run, additional controls over a normal BLAST result appear – the PSIBLAST Threshold can be changed again, or individual sequences can be added or removed from the selection by ticking the box in the first Summary Table column. Go to Step 2 if required The View Threshold limit button jumps the view down the table so you can see the cut off. 55 Sequence Searching and Alignments EBI Bioinformatics Roadshow Controls to download a checkpoint file or PSSM only appear after the second iteration. Instruction: Try running your own PSI-BLAST search using the provided sequence: Search against the UniProtKB/Swiss-Prot database only – if you search against the full UniProt Knowledgebase it will take a very long time! Make sure the PSI-BLAST program is selected Leave the parameters set at their defaults For the moment, stop after the first run 56 Sequence Searching and Alignments EBI Bioinformatics Roadshow Question 18: Looking at the first run (normal BLAST results), how many sequences score above our default threshold of 1.0e-3? Instruction: For the second iteration you can choose which sequences to include in the PSSM generation. Untick the top scoring sequence (simply because it scores so much better than the other results – you wouldn’t necessarily normally do this) Leave everything else set to defaults Click the ‘run next iteration’ button Question 19: Looking at the second iteration, how many sequences now score above our default threshold of 1.0e-3? (Hint: use the View Threshold Limit button). What is a likely explanation? Question 20: Have any new sequences been scored well enough to appear in our results? 57 Sequence Searching and Alignments EBI Bioinformatics Roadshow Filters Filtering can eliminate statistically significant but biologically uninteresting reports from the BLAST output by masking out various segments of the query sequence for regions which are non-specific for sequence similarity searches. This leaves the more biologically interesting regions of the query sequence available for specific matching against database sequences. For example, it may be desired to mask acidic, basic or proline-rich segments of a protein that would otherwise yield overwhelming amounts of uninteresting, non-specific matches against a wide array of protein families. The SEG program (Wootton and Federhen, 1993) masks low compositional complexity regions, while XNU (Claverie and States, 1993) masks regions containing short-periodicity internal repeats. SEG+XNU will combine the above two. The DUST program by Tatusov and Lipman can only be used with DNA searches and will mask simple repeats in DNA/RNA sequences. Instruction: Perform a FASTA search against the UniProtKB/Swiss-Prot database for the filtertest sequence that the demonstrator has provided. Make sure to select more options and set the Histogram display to YES You could also change the Expectation upper value to 0.001 to help make the results clearer To see the histogram, go to the Tool Output tab in the results Question 21: Describe how the observed vs expected histogram looks? What does this mean? How many results have an alignment with an expect score better than 0.001? Instruction: Repeat the search, but this time use the SEG filter from the more options parameters. Make sure that the Histogram display is still set to YES, and expectation value to 0.001 if you want to clearly compare. 58 Sequence Searching and Alignments EBI Bioinformatics Roadshow Question 22: Now how does the observed vs expected histogram look? How many results have an alignment with an expect score better than 0.001? Homologous Over-Extension (HOE) Iterative search strategies using profiles (ie the PSSM in PSI-BLAST) might help increase the sensitivity of a search, however while the aim is to have the profile reflect areas of interest (a domain for example) there is a danger that it will be contaminated with information that is not relevant to your query. Low complexity regions are one example of this, but these can be fixed with the use of filters. Another cause of contamination that was recently described is Homologous OverExtension (HOE). HOE can occur in a profile based alignment when the alignment region picks up a portion of sequence that is not biologically relevant to our query but that is conserved in other sequences brought back by the search. The influence of this region on the scoring matrix can be such that the alignment region extends even further beyond the domain of interest. This can even begin to cover a domain that is not present in the query sequence, once this happens the weighting of the scoring matrix can influence the alignment so much that sequences not at all biologically related to the query start to be found as significant, resulting in an increase in false-positives. Ideally this is prevented by careful selection of which sequences to include in the generation of the PSSM and making sure that they do not have other domains near the boundaries of the alignment that might cause alignment extension – our functional prediction page might help with this. But as this is a manual method, and domain information might not be present in the functional predictions, we have created a method to automatically reduce the likelihood of HOE occurring by masking boundaries at the edge of the original alignment. At the moment this method only applies to PSISearch – a tool that combines sensitive Smith-Waterman based local alignment with the PSI-BLAST profile construction strategy, but it can be enabled by toggling the option ‘HOE region masking’ to yes (which is the default setting). 59 Sequence Searching and Alignments EBI Bioinformatics Roadshow Vector Contamination Another reason you might not get the results you are expecting is due to vector contamination – a common problem if your sequence is fresh from the sequencer. One way to check for this problem if you suspect something is to search against a specialist dataset containing vector sequences only – at the EBI the EMVEC database does exactly this, and there is an NCBI mode to perform this role. 60 Sequence Searching and Alignments EBI Bioinformatics Roadshow Question 23: A student has given you two sequences and they have forgotten whether they have already trimmed them for vector contamination. Use the BLAST tools at EBI to determine whether they have vector contaminants or not. Sequence 1: Sequence 2: 61 Sequence Searching and Alignments EBI Bioinformatics Roadshow Multiple Sequence Alignment We’ve already seen how we can apply rigorous algorithms to align a pair of sequences, but what happens when you want to align more than two sequences? This is where multiple sequence alignment (MSA) comes in. Ideally, a multiple alignment would carry out rigorous alignments between every possible combination of sequences, and then use this Sequences Time information to optimise a final alignment between all of the sequences. Unfortunately this Weighted 2 1 second Sum of Pairs method is incredibly computationally demanding! 3 150 seconds As a result, we have to use heuristics again to bring down alignment times to something that is viable. 4 6.25 hours One method is called progressive alignment. Here subsets of the alignments are carried out and then fixed, to which further alignments take place. This builds up the multiple alignment in a tree fashion. 5 39 days 6 16 years 62 Sequence Searching and Alignments EBI Bioinformatics Roadshow ClustalW ClustalW is an example of a progressive/tree based multiple sequence alignment. It performs a quick pairwise alignment of the sequences before fixing the highest scoring aligned pair and treating them as a single sequence. Other sequences are then aligned to this and fixed in turn, building up a progressive alignment. A guide tree is the term used for the tree created as part of a progressive alignment process, and is used to help order and arrange sequences to be added to a multiple sequence alignment. This is not a phylogenetic tree! A very common mistake is for people to use a guide tree as a phylogenetic tree. This works very quickly, but has some drawbacks, especially if the highest scoring pair are badly aligned, as this alignment error will propagate through the rest of the alignment. ClustalW at the EBI can be found in the sequence analysis section of the tools drop-down menu. Instruction: Navigate to the ClustalW page You should see something like the following: 63 Sequence Searching and Alignments EBI Bioinformatics Roadshow The layout is similar to the pariwise alignment tools we looked at earlier, with an input section and a parameters section. As usual, information about each parameter can be found by clicking the links above each parameter. Key options are described below. Alignment Type ClustalW has the option of performing ‘slow’ or ‘fast’ initial alignments – slow is already quite quick so choose this in most cases. Matrix The comparison matrix to be used to score alignments Gap open Penalty for the first residue in a gap No end gaps Exclude end gaps Gap extension Penalty for each additional residue in a gap Gap distances ClustalW has an additional gap separation penalty No end gaps When set to no this ignores the gap separation penalty at the ends of the alignment 64 Sequence Searching and Alignments EBI Bioinformatics Roadshow Iteration Iteration type Numiter Maximum number of iterations to run Clustering Neighbour Joining is the default clustering option, but UPGMA is available which might help with very large numbers of sequences Output formats What format you want the output file to be in Output order Here you can choose to keep the original input order or to order by alignment 65 Sequence Searching and Alignments EBI Bioinformatics Roadshow Results When your job has finished, you should see the following: Like other tools in our framework, there are several tabs to switch between different results pages. Alignments This page shows the alignment, along with a button to download/show the alignment text file, and another button to colour the sequences according to their physico-chemical properties: Colour Property Residues Red Small (small+ hydrophobic (incl.aromatic -Y)) AVFPMILW Blue Acidic DE Magenta Basic - H RK Green Hydroxyl + sulfhydryl + amine + G STYHCNGQ Grey Unusual amino/imino acids etc Others 66 Sequence Searching and Alignments EBI Bioinformatics Roadshow The key is similar to that which we’ve seen before for other alignment results, however all the sequences are lined up together vertically, and consensus symbols are displayed at the bottom of the columns . Gaps in sequence are displayed with a ‘–‘ character. Where all sequences have the same residue in a column a ‘*’ character is displayed beneath the column. Where similar residues line up there is a ‘:’ character. Where less well conserved substitutions are made they are marked with a ‘.’. Result Summary The result summary page lists the files that the program produces and displays the scores table from ClustalW, which lists the alignment scores for each pair of sequences used to make up the multiple alignment. There is a button to launch Jalview as well. Jalview Jalview is a standalone multiple sequence alignment viewer that allows for more useful viewing than simply looking at the text output of a ClustalW alignment. It can be downloaded and run on its own, however at the EBI we have incorporated an applet version of it into the website, so all you need to do is click the Jalview button on the results page and, assuming java is set up correctly on your machine, Jalview should eventually load with your multiple sequence alignment all ready for viewing. 67 Sequence Searching and Alignments EBI Bioinformatics Roadshow Jalview is quite a powerful tool, with more options than we can go into in this document, however full documentation can be found at the Jalview homepage: http://www.jalview.org/ The graphs under the alignment represent various properties: Conservation measures the number of conserved physic-chemical properties for each column Quality measures the likelihood of observing mutations in a particular column – a high score suggests there are no mutations, or that mutations found are favourable as given by the BLOSUM 62 matrix Consensus shows the most common residue per column and the percentage of alignments that contain this residue, by default gaps are included in this calculation. Guide Tree The next tab is the guide tree. 68 Sequence Searching and Alignments EBI Bioinformatics Roadshow This tab displays any guide trees produced by the MSA tool. Please not that this is NOT a phylogenetic tree! You can download the data for the tree via the button here, or from the link in the Result Summary tab. 69 Sequence Searching and Alignments EBI Bioinformatics Roadshow Submission Details This tab contains all the information about how your job was submitted to the servers, including the command line run and all the parameters. This is very useful if you are wanting to replicate a job on a local machine, and the information on this page is also useful to us in Support if you need help with a problem. Submit Another Job This isn’t really a tab, but takes you back to the start of the form so you can submit a new job. Instruction: Try running your own ClustalW alignment using the provided sequences: You can use email, but the job should be quick enough to run interactively Leave the parameters set at their defaults Turning on colours may make it easier to see regions of similar properties, or you can use Jalview to display the alignment 70 Sequence Searching and Alignments EBI Bioinformatics Roadshow Question 24: This example includes the two sequences we tried to align earlier in the roadshow. Does the multiple alignment give any insight into the result we achieved before? So we’ve tried a fairly simple (and small) multiple sequence alignment. The next few alignments with ClustalW replicate the errors that people ask us for help with, so you know what to do if you see them! Question 25: Perform a ClustalW alignment using the file ‘Problem_MSA1.fsa’ What is the error message shown? What is wrong with our input that caused this error? Question 26: Perform a ClustalW alignment using the file ‘Problem_MSA2.fsa’ What is the error message shown? What is wrong with our input that caused this error? 71 Sequence Searching and Alignments EBI Bioinformatics Roadshow Question 27: Perform a ClustalW alignment using the file ‘Problem_MSA3.fsa’ What is the error message shown? What is wrong with our input that caused this error? Question 28: Perform a ClustalW alignment using the file ‘Problem_MSA4.fsa’ What is the error message shown? What is wrong with our input that caused this error? 72 Sequence Searching and Alignments EBI Bioinformatics Roadshow Other Multiple Sequence Alignment Tools While ClustalW is by far the most popular MSA tool currently, it has a number of drawbacks, notably in the danger of propagating errors from the initial alignments throughout the whole alignment or the way it deals with unusual alignments. Nonetheless its speed and wide usage make it a useful tool. Clustal Omega Clustal Omega is the latest tool from the Clustal authors. It uses a number of new techniques to significantly improve alignments over ClustalW including seeded guide tree generation and HMMHMM alignments. Other MSA tools are available at the EBI to enable you to perform more accurate alignments or at least to compare the results between different alignments. Some of these are mentioned below: T-Coffee T-Coffee is a tree-based variant of the COFFEE tool, which aims to keep some of the accuracy while enabling it to be run on a viable timescale. It still has some high demands on computer hardware however, and large jobs can take a very long time to run! MUSCLE MUSCLE is another progressive alignment tool, but goes about things in a much cleverer way than ClustalW with the result that accuracy is claimed to be higher for the same or better speed. MAFFT MAFFT uses Fast Fourier Transforms to perform accurate and fast alignments. Kalign Kalign uses an approximate string matching algorithm to estimate sequence distances very rapidly, concentrating on local regions rather than globally aligning, and is the fastest algorithm we offer for large numbers of sequences. The tools can all be found either from the drop down menus or from the Multiple Sequence Alignments page at the EBI (www.ebi.ac.uk/Tools/msa): 73 Sequence Searching and Alignments EBI Bioinformatics Roadshow The way the tools are used is very similar to the way ClustalW is used. You should be able to launch Jalview for most of our MSA tools, however if the option is missing you can launch Jalview from another tool and paste the alignment from your tool into it, to view the alignment. 74 Sequence Searching and Alignments EBI Bioinformatics Roadshow Instruction: Try running your own alignments using the provided sequences and trying out different MSA tools: Choose from any or all of Clustal Omega, MUSCLE, MAFFT, T-Coffee, Kalign See if you can tell any difference in running speed (you might not be able to – this is a very short alignment) Compare the alignment results with other tools including ClustalW You might find it easier to use Jalview to compare several alignments You can cut/paste sequences in Jalview to re-order them Question 29: Note any comments you have about the different alignment tools: 75 Sequence Searching and Alignments EBI Bioinformatics Roadshow WebPRANK So far the tools we’ve looked at for MSA tend towards roughly similar behaviours. It has been speculated that this might be because they are usually benchmarked against only a few specific datasets, and thus they tend to be optimised towards high scoring results for those tests. Also they tend to favour multiple independent deletions over insertions, leading to sequences that shrink in length over evolution, which isn’t a view backed up by evolutionary evidence. PRANK is a tool which tries to address these shortcomings by using phylogenetic information to keep track of deletions as they occur through the sequence evolution. PRANK was developed by the Goldman group (and Ari Löytynoja in particular) at the EBI, so has its own page as part of the Goldman research group http://www.ebi.ac.uk/goldman/. pages, which can be found at Instruction: Navigate to http://www.ebi.ac.uk/goldman-srv/webprank/ 76 Sequence Searching and Alignments EBI Bioinformatics Roadshow As you can see, it looks a little different from our general sequence analysis tools. It should open with the Sequence input and alignment section open, and it is here that you can paste or upload your sequence. This is also where the Start alignment button is located. To access the options for changing parameters, you have to click on the links below the input section. The previously open section will contract and the new section will open and allow you to view or make changes. You can also retrieve previously submitted jobs, or use the wePRANK tools to view alignments from another source (or that were previously saved). Results Once the job has run you have several options. You can open the results in the browser, open them in the webPRANK viewer, or download the results. The job-ID is also listed should you want to note it down to retrieve at another date. The webPRANK viewer allows you to view the alignment interactively, as well as the phylogentic information that has helped inform the alignment. There is also a reliability score which allows you to remove sites with lower reliability, either based on the currently selected node or on the lowest score. This will mask portions of the alignment, allowing you to export just the higher reliability sections. 77 Sequence Searching and Alignments EBI Bioinformatics Roadshow Instruction: Try running your own webPRANK alignment using the provided sequences. Input the sequences in the top section Have a look at the different options available in the other sections But keep to defaults for this run When the job is finished you can view the results with several methods, try the webPRANK viewer Question 30: How does the alignment in webPRANK compare with ClustalW? How long is the alignment? What are the likely reasons for this? 78 Sequence Searching and Alignments EBI Bioinformatics Roadshow Getting HELP Read the database Documentation Frequently Asked Questions: http://www.ebi.ac.uk/help/faq.html 2can Support Portal: http://www.ebi.ac.uk/2can/ EBI Support: http://www.ebi.ac.uk/support/ Hands-on training programme: http://www.ebi.ac.uk/training/handson/ Related articles from the EBI A new bioinformatics analysis tools framework at EMBL-EBI [http://dx.doi.org/10.1093/nar/gkq313] The European Bioinformatics Institute’s data resources [http://dx.doi.org/10.1093/nar/gkp986] Web services at the European Bioinformatics Institute-2009 [http://dx.doi.org/10.1093/nar/gkp302] The Universal Protein Resource (UniProt) in 2010 [http://dx.doi.org/10.1093/nar/gkp846] The IntAct molecular interaction database in 2010 [http://dx.doi.org/10.1093/nar/gkp878] The Gene Ontology in 2010: extensions and refinements [http://dx.doi.org/10.1093/nar/gkp1018] The Proteomics Identifications database: 2010 update [http://dx.doi.org/10.1093/nar/gkp964] InterPro: the integrative protein signature database [http://dx.doi.org/10.1093/nar/gkn785] Reactome knowledgebase of human biological pathways and processes [http://dx.doi.org/10.1093/nar/gkn863] Further reading Needleman, S. B. and Wunsch, C. D. (1970) A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48, 443–453 Smith, T. F and Waterman, M. S. (1981) Identification of common molecular subsequences. J. Mol. Biol. 147, 195–197 Pearson, W. R. and Lipman, D. J. (1988) Improved tools for biological sequence comparison. Proc. Natl Acad. Sci. U. S. A. 85, 2444–2448 Ning, Z., Cox, A. J. and Mullikin, J. C. (2001) SSAHA: a fast search method for large DNA databases. Genome Res. 11, 1725–1729 Kent, J. (2002) BLAT – the BLAST-like alignment tool. Genome Res. 12, 656–664 Dayhoff, M. O., Schwartz, R. M. and Orcutt, B. C. (1978) A model for evolutionary change in proteins. in Atlas of Protein Sequence and Structure, (Ed. Dayhoff, M. O.) Vol. 5, pp. 345–352 (National Biochemical Research Foundation) Henikoff, S. and Henikoff, J. G. (1992) Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. U. S. A. 89, 10915–10919 Altschul,S.F., Warren,G., Webb,M., Eugene,W.M. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol. 215:403–410 Altschul,S.F., Madden,T.L., Schäffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25(17):3389-402. Lopez,R., Silventoinen,V., Robinson,S., Kibria,A. and Gish,W. (2003) WU-Blast2 server at the European Bioinformatics Institute. Nucleic Acids Res. 31(13):3795-8 . Mackey,A.J., Haystead,T.A. and Pearson,W.R. (2002) Getting more from less: algorithms for rapid protein identification with multiple short peptide sequences. Molecular and Cellular Proteomics 1(2):139-147. 79 Sequence Searching and Alignments EBI Bioinformatics Roadshow Brown,N.P., Leroy,C. and Sander,C. (1998) MView: a web-compatible database search or multiple alignment viewer. Bioinformatics 14(4):380-381. Thompson,J.D., Plewniak,F., Thierry,J.C. and Poch,O. (2000) DbClustal: rapid and reliable global multiple alignments of protein sequences detected by database searches. Nucleic Acids Res. 28(15):2919-2926. Mickael Goujon, Hamish McWilliam, Weizhong Li, Franck Valentin, Silvano Squizzato, Juri Paern and Rodrigo Lopez (2010) A new bioinformatics analysis tools framework at EMBL–EBI. Nucleic Acids Res. 31(13):3795-8 . Mileidy W. Gonzalez and William R. Pearson (2010) Homologous over-extension: a challenge for iterative similarity searches. Nucleic Acids Res. 2010 April; 38(7): 2177–2189 Appendix Nucleotide codes Code Meaning Etymology Complement Opposite A A Adenosine T B T/U T Thymidin/Uridine A V G G Guanine C H C C Cytidine G D K G or T Keto M M M A or C Amino K K R A or G Purine Y Y Y C or T Pyrimidine R R S C or G Strong S W W A or T Weak W S B T or G or C not A (B is next) V A V A or G or C not T/U (V is next) B T/U H A or T or C not G (H is next) D G D A or T or G not C (D is next) H C X/N A or T or G or C any N . . not A or T or G or C . N 80 Sequence Searching and Alignments EBI Bioinformatics Roadshow Amino acid codes Single letter code 3-letter code Name A Ala Alanine C Cys Cysteine G Gly Glycine H His Histidine I Ile Isoleucine L Leu Leucine M Met Methionine P Pro Proline S Ser Serine T Thr Threonine V Val Valine F Phe Phenylanine N Asn Asparagine R Arg Arginine Y Tyr Tyrosine D Asp Aspartic acid E Glu Glutamic acid K Lys Lysine Q Gln Glutamine W Trp Tryptophan B Asx Asp or Asn Z Glx Glu or Gln X Any 81 Sequence Searching and Alignments