Proposta di ricerca: Biological activity of protein molecules depends
advertisement

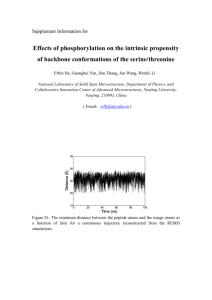
Proposta di ricerca: Biological activity of protein molecules depends on their structure and internal dynamics, including collective slow protein motions resulting from structural fluctuations. Normal mode analysis represents a powerful and cheap way to study such motions, but they cannot catch phenomena out of equilibrium, such as movements of protein fragments induced by the presence of ligands, or triggered by signal transduction processes. These phenomena can be simulated by molecular dynamics (MD), however robust and efficient tools are required for postprocessing the huge amount of data generated. In this context, Dr. Caliandro with his coworkers developed a new tool, based on internal descriptors (backbone dihedral angles) [1]. It overcomes the limitations of the existing ones and proved very effective in identifying hinge motions and hot spot residues [1,2,3]. It has been also used to explain flexibility features observed in intrinsic disordered proteins upon ligand binding [4]. On the other hand, Dr. Lesyng with his coworkers developed a multivariate autoregressive model using selected degrees of freedom, which was able to capture causal relations in molecular structural changes, giving valuable information to interpret the mechanisms of functioning of biomolecular systems [5,6]. Also a web service capable to indicate dynamic proteins domains from MD simulation data is being currently developed [7]. Within this project we plan to combine the mentioned above approaches, based on directional statistics of dihedral descriptors with the dynamic domain identification and Granger-type causality analyses, to achieve a deeper understanding of the dynamics of complex biomolecules, and characterizing the out-of-equilibrium functional movements of protein domains. As a further step, we plan to develop a comprehensive tool which would be able to process MD data or ensemble of experimentally determined protein conformations to identify dynamic domains, residues which serve as hinge points, and interfacial regions in proteins. The use of internal coordinates should allow differentiating motions consisting in fluctuations around a given configuration from those representing a transition between different configurations (in this case the system experiences a conformational change). New algorithms for clustering protein conformations sampled during MD could be consequently envisaged. They will be based on the identification of dynamic domains [7] and on recent algorithms for structural comparison [8]. Classification of protein conformations based on their internal dynamics will be used to facilitate the protein crystal structure solution. The more frequent method for protein crystal solution (68% of existing structures) is molecular replacement (MR), where the correct position of a rigid model of an homologous protein in the crystal cell is searched by fitting diffraction intensities. Novel and more efficient MR procedures have been recently developed [9,10], which still require small deviations between model and target structures. Crystallographic methods developed by the Dr. Caliandro group will be combined to computational modelling skills and algorithms developed by the Dr. Lesyng group to push forward actual limits of MR procedures, according to the following strategy: 1) generate a structural model by homology modelling, by using the target sequence 2) execute molecular dynamics simulations on the generated model 3) a) identify the dynamic domains of the model, by using tools developed within this project b) sample their more frequent conformations and discard very flexible linkers among domains 4) fit the dynamic domains in their selected conformations as rigid bodies in the crystal cell - in fact, existing MR procedures are able to fit several different models in the unit cell, provided they are large enough to produce a sensible effect on the used cost functions. This procedure aims at introducing flexibility of the search model in the MR procedure, to account for structural differences with the target structure. At different stages of the pipeline the information on the mutual orientation of alphahelices will be used as constraint. It is supplied by a recent algorithm which identifies alpha-helix directly from diffraction patterns [11]. References: [1] R. Caliandro, G. Rossetti, P. Carloni "Local fluctuations and conformational transitions in proteins", J. Chem. Theory Comput., 2012 8 4775; [2] G. Rossetti, X.J. Cong, R. Caliandro et al. "Common Structural Traits across Pathogenic Mutants of the Human Prion Protein and Their Implications for Familial Prion Diseases", J. Mol. Biol. 2011 411 700; [3] R. Caliandro, G. Nico, G. Tiscia et al., "Structural analysis of protein Z gene variants in patients with foetal losses", Thromb. Haemost. 2013 110 534; [4] D. Dibenedetto, G. Rossetti, R. Caliandro et al. "A molecular dynamics simulation-based interpretation of nuclear magnetic resonance multidimensional heteronuclear spectra of alphasynuclein•dopamine adducts", Biochem. 2013 52 6672; [5] A. Gorecki, J. Trylska, B. Lesyng "Causal relations in molecular dynamics from the multi-variate autoregressive model", Europhys. Lett., 2006 75 503; [6] P. Daniluk, M. DziubiDski, B. Lesyng et al. "From experimental, structural probability distributions to the theoretical causality analysis of molecular changes", CAMES, 2012 19 257; [7] M. Dziubinski, P. Daniluk, B. Lesyng "ResiCon: a web server for the identification of dynamic domains, hinges and interfacial regions in proteins", in preparation; [8] P. Daniluk, B. Lesyng "A novel method to compare protein structures using local descriptors", BMC Bioinform. 2011 12 344; [9] R. Caliandro, B. Carrozzini, G.L. Cascarano et al. "Molecular replacement: the probabilistic approach of the program REMO09 and its applications" Acta Cryst. 2009 A65 512; [10] B. Carrozzini, G.L. Cascarano, G. Comunale et al. "The use of VLD (vive la difference) in the molecular-replacement approach: a pipeline", Acta Cryst. 2013 D69, 1038; [11] R. Caliandro, D. Dibenedetto, G.L. Cascarano et al. "Automatic alpha-helix identification in Patterson maps", Acta Cryst. 2012 D68, 1;