Supplemental material jcp_reviewed
advertisement

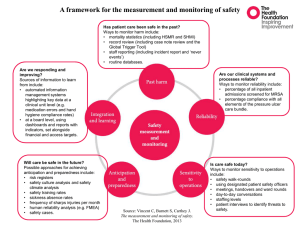
Supplemental Material Communication: Vibrational spectroscopy of atmospherically relevant acid cluster anions – bisulfate versus nitrate core structures Tara I. Yacovitch, Nadja Heine, Claudia Brieger, Torsten Wende, Christian Hock, Daniel M. Neumark, Knut R. Asmis FULL CITATION FOR GAUSSIAN 09 Gaussian 09, Revision C.01, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery Jr., J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Keith, T.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J.; Gaussian, Inc.: Wallingford CT, 2010. 1 SIMULATED SPECTRA 1a 1b 1c FIG. S1. Comparison between experimental IRMPD spectrum for cluster 1 and simulated IR intensities calculated at the B3LYP/6-311++G(2df,2pd) level of theory. Simulated line spectra are convoluted with a Gaussian of FWHM=15 cm-1. Theoretical isomers are labeled according to their relative energy, which includes ZPE. 2 1d 2a 2b 2c 2d 2e 2g FIG. S2. Comparison between experimental IRMPD spectrum for cluster 2 and simulated IR intensities calculated at the B3LYP/6-311++G(2df,2pd) level of theory. Simulated line spectra are convoluted with a Gaussian of FWHM=15 cm-1. Theoretical isomers are labeled according to their relative energy, which includes ZPE. 2j 2l 3 3a 3b 3d 3f 3g FIG. S3. Comparison between experimental IRMPD spectrum for cluster 3 and simulated IR intensities calculated at the B3LYP/6-311++G(2df,2pd) level of theory. Simulated line spectra are convoluted with a Gaussian of FWHM=15 cm-1. Theoretical isomers are labeled according to their relative energy, which includes ZPE. 3h 3i 4 4a FIG. S4. Comparison between experimental IRMPD spectrum for HSO 4¯ and simulated IR intensities calculated at the B3LYP/6-311++G(2df,2pd) level of theory. Simulated line spectra are convoluted with a Gaussian of FWHM=15 cm-1. 5 PEAK POSITIONS AND ASSIGNMENTS TABLE SI. Summary of experimental peaks and assignments. Selected unscaled harmonic frequencies, simulated at the B3LYP/6-311++G(2df, 2pd) level, are given next to an approximate description of the normal mode. Experimental peak (cm-1) Theoretical peak (cm-1) Theoretical structure Approximate normal mode description.a Cluster 1: HSO4¯·HNO3 890 900 1a 942 990 1b 1031 1186 944 1164 1316 1a 1b 1b (H2SO4): symmetric SOH wags (HNO3): NO3 symmetric stretch, (HSO4¯): symmetric SO3 stretch (H2SO4): symmetric SOH wags (HSO4¯): SOH bend and S=O stretch (HSO4¯): SOH bend and S=O stretch, (HNO3): NOH bend and N-O stretch / out-of-phase version of the same vibration (HNO3): NOH bend (NO3¯): N=O stretch, H2SO4: SOH bends (HNO3): NOH bend and N=O stretch 1310 / 1338 1400 1478 1483 1462 1621 1652 Cluster 2: HSO4¯·H2SO4·HNO3 590 575 875 911 934 974 1045 1030 1155 1168 / 1b 1b 1a 1b 2a 2a 2a 2a 2a 2a 2a 2a (HSO4¯): SO2 bend (H2SO4): antisymmetric S-OH stretch (HNO3): symmetric NO3 stretch (HSO4¯): symmetric SO3 stretch (H2SO4): symmetric SO2 stretch, (HSO4¯): SOH bend and S=O stretch / (HSO4¯): antisymmetric SO2 stretch (HSO4¯): SOH bend and S=O stretch, SO and NO stretches on other moieties (HSO4¯): SOH bend, (HNO3): symmetric N-OH stretch (HNO3): antisymmetric NO3 stretch, (HSO4¯): SOH bend (H2SO4): SOH wags (HNO3): NOH bend (HNO3): NOH bend and N=O stretch 3a 3a (HNO3): symmetric NO3 stretch (H2SO4): symmetric SO2 stretch / 1171 / 2a 1224 1206 2a 1300 1318 2a 1338 1350 2a 1399 1388 1443 1427 1660 1687 Cluster 3: NO3¯·H2SO4·HNO3 945 978 1116 1185 6 1297 1322 3a 1349 1339 3a 1399 3a 1412 / 1415 / 3a 1650 Ion 4: 1131 1198 1244 1689 HSO4¯ 1137 1224 1280 3a (HNO3): NOH bend and antisymmetric NO3 stretch, NO and SO stretches on other moieties (H2SO4): antisymmetric SO2 stretch and SOH wags, (HNO3): antisymmetric stretch (NO3¯): antisymmetric stretch, stretches and wags on other moieties / (NO3¯): antisymmetric stretch, stretches and wags on other moieties (HNO3): NOH bend and N=O stretch 4a 4a 4a SOH bend Antisymmetric S=O stretch SOH bend and S=O stretch a. Descriptions of cluster normal modes are given in terms of the moieties where vibration occurs. A normal mode involving motion of two moieties would be described thusly: (Moiety1): description of the vibrations of moiety 1, (Moiety2): description of the vibrations of moiety 2. A forward slash “/” is used to separate different normal modes. 7 THERMODYNAMIC CALCULATIONS TABLE SII details relative energetics of structures 1a and 1b with and without inclusion of zero point energy. Thermodynamic corrections for enthalpy and entropy are also included at two different conditions: 298.15K / 1atm and 15.0K / 4·10-4atm. At trap temperatures, inclusion of enthalpy or free energy does not much impact the realtive energetics and even at 298K, structure 1a remains energetically favorable by all measures. However, at STP, the free energy of 1b lies only 14.0 kJ/mol above 1a. Not included in this calculation is the entropy contribution from internal rotations. We expect this contribution to be negligible for 1a but significant for 1b: both the nitric acid and free OH bond in 1b are free to rotate while the presence of two H-bonds limits such motion in 1a. We estimate that this contribution could further decrease the free energy by around 2 kJ/mol for 1b at 298.15K (method of Pitzer and Gwinn1-3). Thus, entropy effects lower the relative free energy of 1b at higher temperatures. TABLE SII. Relative energies (kJ/mol) for the two lowest energy structures of cluster 1 calculated at the B3LYP/6-311++G(2df,2pd) level.a The relative electronic energy (Eelec) is shown along with corrections due to zero point energy (ZPE), enthalpy (ΔH) and free energy (ΔG). The dissociation energy of the clusters relative to their geometry optimized fragments is also calculated. 1a 0.00 0.00 0.00 0.00 0.00 0.00 101 Eelec Eelec + ZPE Eelec + ΔH15K Eelec + ΔG15K Eelec + ΔH298K Eelec + ΔG298K D0 1b 28.3 24.3 24.4 24.2 27.0 14.0 175 a. Thermodynamic quantities were calculated at the following conditions: 15 K and 4.0·10 -4 atm; 298.15K and 1.00 atm. D0 includes ZPE. ΔH and ΔG include internal energies (ZPE, rotational and translational) but do not account for internal rotations within the cluster. 8 POTENTIAL ENERGY SURFACE SCANS A potential energy surface scan was done to investigate the rotation of the HNO 3 moiety in structure 1b by scanning the S-O-O-N dihedral angle (Fig. S5). At ~100° there is a small barrier before acessing the deep attractive well of the second hydrogen bond (a structure which would converge to 1a), and at ~330° there is a high barrier due to repulsion from a bisulfate oxygen. The lowest of these two barriers is ~2.5 kJ/mol and corresponds to a kT temperature of 297 K. This is significantly higher than the trap temperature and likely higher than the evaporating droplet temperature in the He filled ion guide. Thus, we expect that most clusters formed on this shallow HSO4¯·HNO3 surface can relax during ion guide or ion trap collisions down to structure 1b, but perhaps not to the minimum energy structure 1a. FIG. S5. Rigid scan of the SOON dihedral angle of the HSO4¯·HNO3 cluster 1b. Calculations were done at the B3LYP/6-311++G(2df,2pd) level. Energies in kJ/mol are relative to the 1a global minimum and do not include zero point energies. 9 REFERENCES (1) Pitzer, K. S.; Gwinn, W. D. J. Chem. Phys. 1942, 10, 428. (2) Pitzer, K. S. J. Chem. Phys. 1946, 14, 239. (3) Janz, G. J. Thermodynamic Properties of Organic Compounds: Estimation Methods, Principles and Practice; Revised ed.; Academic Press: New York, 1967. 10