emi412292-sup-0003-si
advertisement

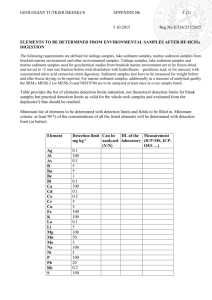
1 Supplementary Material and Methods 2 Experimental setup and sample collection 3 Surface sediment (top 20 cm) was collected from the mouth of the Plym Estuary 4 (50°22’17.22’’N, 04°06’34.45’’W) in the South West of the UK. The sediment was sieved 5 through a 1mm mesh to remove large fauna, stones and other debris. In order to 6 contaminate the sediment with oil, 375 g was air dried before 20 g of IFO-180 crude oil was 7 dissolved in 100 mL hexane and mixed with the dry sediment. The oil-contaminated 8 sediment was incubated overnight with continuous airflow, and after the hexane had 9 evaporated was mixed with 1125 g of fresh sediment. The contaminated sediment was 10 maintained for 2 months submerged in a continuous flow seawater aquarium at 15°C and in 11 the dark before experimental setup. Oil-contaminated sediment (450 g) was transferred to 12 individual plastic tubes (length 18 cm, diameter 6 cm) that were sealed at one end 13 (Supplementary Figure 1). The tubes were maintained submerged in continuous flow 14 seawater aquaria at 15°C and in the dark. After 7 days, 2 adult H. diversicolor with the same 15 length and weight were added to 3 experimental cores and 3 cores were used as controls 16 (i.e. no bioturbation). The polychaetes formed gallery burrows within 2-3 days and remained 17 active throughout the experimental period (Supplementary Figure 1). 18 After 30 days, a single sediment sample (4.75 cm3) was taken from each core 19 containing H.diversicolor from a burrow-enriched area of the sediment. The sediment was 20 collected 5 cm below the sediment water interface using plastic mini-cores (0.95 cm2 surface 21 area, 5 mL syringe with the end cut off), these samples are referred to as “burrows”. In cores 22 without H.diversicolor sediments were sampled in the same way and are referred to as “un- 23 bioturbated sediments”. The sampled sediment was transferred into 2 mL micro-centrifuge 24 tubes and stored at -80oC until further analysis. 25 26 DNA extraction 27 DNA was extracted using the DNeasy extraction kit (Qiagen) with minor modifications. 28 Sediment (0.25 g) was weighed into tubes containing glass beads (0.5 g, 100–300 μm 1 29 diameter, MPBIO lysing matrix B). ATL lysis buffer (Qiagen) (600 μL) was added to the 30 tubes. The tubes were then placed in a bead beater (Mini-Bead Beater 1, BioSpec) and 31 beadbeated at maximum speed (48 rpm) for 1 minute. The tubes were incubated at 56°C for 32 10 minutes before being beadbeated for another 1 minute and a final incubation at 56°C for a 33 further 10 minutes. The tubes were spun (10,000 rpm, 30s), and to remove humic 34 acids/inhibitors the supernatant added to a QIA-shredder column (Qiagen) containing 0.1 g 35 Polyvinylpolypyrrolidone (PVPP), incubated at room temperature for 10 minutes and finally 36 spun (12,000 g, 30s). The rest of the protocol was carried out according to the 37 manufacturer’s instructions. The DNA was re-suspended in elution buffer and quantified 38 using a Nanodrop® ND-1000 spectrophotometer (Thermo-scientific). 39 40 RNA extraction and cDNA synthesis 41 Sediment (0.25g) was weighed into tubes containing 0.5g glass beads (100–300 μm, 42 MPBIO) and 1mL TRI Reagent® (Ambion) before bead beating. Samples were heated at 43 60°C for 10 min before 600 μL of supernatant was transferred to a new tube containing 44 100µl 1-bromo-3-chloro-propane and vortexed. The tubes were centrifuged to separate the 45 organic and aqueous phases before the aqueous phase was transferred to a QIA-shredder 46 column containing PVPP as described above. The filtrate was precipitated with 2-propanol 47 (equal volume) and sodium acetate (1/10 volume) for 1 hr at -20°C before the RNA pellet 48 was isolated and washed by centrifugation. The RNA pellet was resuspended in 100 μL 49 RNAse-free water and further cleaned using the RNeasy kit (Qiagen) according to the 50 manufacturer’s instructions. DNAse treatment was performed using RQ1 RNase-Free 51 DNase (Promega) according to the manufactures instructions. Control PCRs confirmed the 52 presence of only RNA. cDNA generation was performed using an Omniscript RT kit (Qiagen) 53 in accordance with manufacturer’s instructions. 54 2 55 16S rRNA and 18S rRNA transcript quantification 56 For bacteria qPCR, the primers of Suzuki et al (2000) were used (Forward BACT1369F 5´- 57 CGG TGA ATA CGT TCY CGG-3´, Reverse PROK1492R 5´-GGW TAC CTT GTT ACG 58 ACT T-3´) and for eukaryotes those of Zhu et al (2005) (Forward EUK345f 5´- 59 AAGGAAGGCAGCAGGCG-3´, Reverse EUK499r 5´-CACCAGACTTGCCCTCYAAT-3´). 60 qPCR was carried out using the Sensi-FAST SYBR Q-PCR kit (Bioline) in 10µl reactions 61 containing 5 μL of sensi-fast master mix, 0.25 μL of each primer (final concentration 0.4 μM), 62 1 μL of cDNA template and 4 μL nuclease free water. A Qiagen rotor gene3000 (Qiagen) 63 was used to perform the reactions. Cycling conditions were an initial denaturation of 94°C 64 for 3 min, then 40 cycles of 94°C for 10s, annealing for 15s at 59°C for bacteria or 60°C for 65 eukaryotes, elongation and acquisition of fluorescence data at 72°C for 20s. Standard curves 66 for Q-PCR were constructed using known amounts of purified target template generated by 67 PCR amplification of the target gene from genomic DNA from either Escherichia coli or 68 Saccharomyces cerevisiae. Transcript copy numbers were back calculated to water content 69 corrected sediment weights (see below). 70 71 Sediment water content 72 Sediment (0.25g) was sub-sampled from each sample for DNA/RNA analysis and weighed 73 into pre-combusted (550°C for 8 hours) glass petri-dishes. The dishes were placed in a 74 drying oven at 60°C overnight and reweighed. Water content (%) was calculated as: 75 𝑊𝑎𝑡𝑒𝑟 𝑐𝑜𝑛𝑡𝑒𝑛𝑡 (%) = (1 − Dry weight sediment + dish (g) ) × 100 Wet weight sediment + dish (g) 76 77 16S rRNA/18S rRNA amplicon sequencing and bioinformatics 78 Bacteria 16S rRNA and eukaryote 18S rRNA gene amplicon sequencing was carried as 79 previously described (Taylor et al., 2014; Taylor and Cunliffe, 2014). In summary, the V4 80 variable region of the 16S rRNA gene was amplified using the PCR primers 515F and 806R 81 (Caporaso et al., 2011), and the following PCR conditions: 94°C for 3 minutes, followed by 3 82 28 cycles of 94°C for 30 seconds, 53°C for 40 seconds and 72°C for 1 minute, and a final 83 elongation step at 72°C for 5 minutes. The V9 variable region of the 18S rRNA gene was 84 amplified using PCR primers 1391F (Lane, 1991) and EukB (Medlin et al., 1988). The same 85 PCR conditions were used except the annealing temperature was changed to 57°C. 86 Sequencing of amplified 16S rRNA and 18S rRNA genes was performed on an Ion Torrent 87 PGM (Life technologies) according to manufactures instructions. 88 Sequences were analysed using the QIIME software package (Caporaso et al., 2010) 89 as previously described for 16S rRNA (Taylor et al., 2014) using the Greengenes database 90 (DeSantis et al., 2006) and 18S rRNA (Taylor and Cunliffe, 2014) using the SILVA database 91 (Quast et al., 2013) as a reference. In brief, quality filters were used to remove short 92 (<250bp bacteria, <150bp eukaryotes) and low quality reads (average phred score <25). 93 Chimeras were then identified and removed. Operational taxonomic units (OTUs) were 94 defined at 97% similarity and classified against the reference databases. 95 96 Hydrocarbon analysis 97 After 30 days, the H. diversicolor were removed and the experimental cores sacrificed for 98 hydrocarbon analysis. The sediment was homogenised before hydrocarbon analysis using 99 standard protocols. In summary, aliphatic and aromatic hydrocarbons were extracted in 100 hexane/acetone and analysed using GC-FID. PAHs were extracted in 101 hexane/acetone/triethylamine and analysed using GC-MS. 102 103 Statistical analyses 104 Significant differences in hydrocarbon concentrations and Q-PCR results were determined 105 by t-tests using the SPSS statistics software package (IBM). Permutational Multivariate 106 Analysis of Variance (PERMANOVA) was performed with 999 permutations in QIIME using 107 UniFrac distance matrices and OTU tables as inputs to investigate differences in community 108 composition at the OTU level. In order to establish if there were significant differences 109 between burrows and sediment in the relative abundance of specific eukaryote and bacteria 4 110 groups, data were converted from percentages to arcsin values before t-test analysis using 111 SPSS. 112 113 References 114 Caporaso, J.G., Lauber, C.L., Walters, W.A., Berg-Lyons, D., Lozupone, C.A., Turnbaugh, 115 P.J. et al. (2011) Global patterns of 16S rRNA diversity at a depth of millions of 116 sequences per sample. Proceedings of the National Academy of Sciences 15: 4516- 117 4522. 118 Caporaso, J.G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F.D., Costello, E.K. et 119 al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nature 120 Methods 7: 335-336. 121 DeSantis, T.Z., Hugenholtz, P., Larsen, N., M, R., Brodie, E.L., Keller, K. et al. (2006) 122 Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible 123 with ARB. Applied and Environmental Microbiology 72: 5069-5072. 124 Lane, D.J. (1991) 16S/23S rRNA sequencing In Nucleic Acid Techniques in Bacterial 125 Systematics. Stackebrandt, E., and Goodfellow, M. (eds). Chichester, UK: John Wiley & 126 Sons, pp. 115-175. 127 Hamady, M., Lozupone, C., & Knight, R. (2009). Fast UniFrac: facilitating high-throughput 128 phylogenetic analyses of microbial communities including analysis of pyrosequencing and 129 PhyloChip data. The ISME journal, 4(1), 17-27. 130 131 132 Medlin, L., Elwood, H.J., Stickel, S., and Sogin, M.L. (1988) The characterization of enzymatically amplified eukaryotic 16S-like rRNA-coding regions. Gene 71: 491–499. Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P. et al. (2013) The 133 SILVA ribosomal RNA gene database project: improved data processing and web-based 134 tools. Nulceic Acids Research D590-596. 135 Suzuki, M.T., Taylor, L.T., and DeLong, E.F. (2000) Quantitative analysis of small-subunit 136 rRNA genes in mixed microbial populations via 5′-nuclease assays. Applied and 137 Environmental Microbiology 66: 4605-4614. 5 138 Taylor, J.D., and Cunliffe, M. (2014) High-throughput sequencing reveals neustonic and 139 planktonic protist diversity in coastal waters. Journal of Phycology 50: 960–965. 140 Taylor, J.D., Cottingham, S.D., Billinge, J., and Cunliffe, M. (2014) Seasonal microbial 141 community dynamics correlate with phytoplankton-derived polysaccharides in surface 142 coastal waters. ISME Journal 8: 245-248. 143 Zhu, F., Massana, R., Not, F., Marie, D., and Vaulot, D. (2005) Mapping of picoeukaryotes in 144 marine ecosystems with quantitative PCR of the 18S rRNA gene. FEMS Microbiology 145 Ecology 52: 79-92. 146 147 6