Description of HD
advertisement

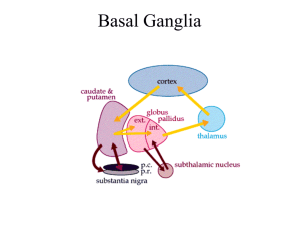
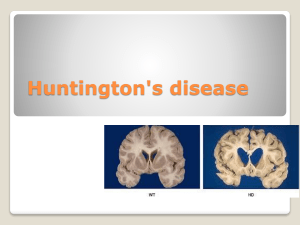
Description of Huntington’s Disease By Erica Metheny Introduction Huntington’s disease is a hereditary neurodegenerative disease that causes chorea along with psychiatric and cognitive symptoms. The average age of symptom onset is 40 years and they typically last for 20 years before the disease becomes fatal. Some of the more common symptoms include chorea, weakness, and depression. The area of the brain that experiences the most damage is the basal ganglia in both direct and indirect pathways. Normal huntingtin protein is involved in synaptic transmission, neuroprotective factors, and BDNF production and transport. All of those functions are affected in people that have Huntington’s disease. The purpose of this document is to describe the neurodegeneration that occurs in Huntington’s disease to students interested in neuroanatomy or brain disorders. Having a basic background in neuroanatomy and genetics will help the reader better understand this document. Symptoms As stated earlier, during all stages of the disease patients experience motor, cognitive, and psychiatric symptoms. In the early stages patients develop fidgeting movements, clumsiness, irritability, impulsivity, depression, and forgetfulness. Moderate stage is characterized by a progression of motor symptoms such as chorea, dystonia (sustained muscle contractions), and slow voluntary movements. Some late stage symptoms include chorea in almost all patients, rigidity, swallowing problems, inability to care for self, depression (50% of patients), mania, memory problems, inability to speak, and comprehension issues. On average patients start to see symptoms around age 40 and they last for approximately 20 years. Patients with Huntington’s disease typically die from pneumonia, choking, or cardiovascular disease. Genetics Huntington’s disease is due to a mutation on a gene located on chromosome four that codes for the huntingtin protein. It is autosomal dominant meaning that someone with Huntington’s disease has a 50% chance of having a child with the disease. The huntingtin gene (htt) has a sequence of 3 mRNA bases (CAG) that are repeated many times. People that have over 40 CAG repeats are guaranteed to have Huntington’s disease. Those with 36-40 repeats fall under the category of reduced penetrance and have a possibility of developing the disease and people with 35 or less repeats will not get Huntington’s. The basic function of the huntingtin protein is cell structure, metabolism, and transport. Mutant proteins stick together and cause the protein to fold into the incorrect shape. This causes the protein to form abnormal clumps inside the neuron (neuronal intranuclear inclusions). The altered form of the huntingtin protein cannot get broken down in the cell and the cells die. Overall, Huntington’s is a genetic disease caused by having more than 40 CAG repeats on chromosome four. These repeats cause a mutant huntingtin gene resulting in disruption of cell structure, metabolism, and transport. Anatomy of Huntington’s Disease The basal ganglia largely controls voluntary movement and undergoes the most damage in Huntington’s. There is a significant amount of tissue damage done to the structures in the basal ganglia before symptoms even start to show. Structures of the basal ganglia include the striatum (including the caudate nucleus), nucleus accumbens, globus pallidus, subthalamic nuclei, and the substantia nigra. The striatum’s main function is to regulate and inhibit movement. The subthalamic nuclei are responsible for inhibiting movement and action selection. The globus pallidus is involved in voluntary movement such as walking and talking. Lastly, the Thalamus is the relay station for sensory and motor signals to the cerebral cortex. The internal globus pallidus has tonic inhibition on the thalamus, meaning that unless there is another signal, the internal globus pallidus in inhibiting the thalamus. There are 2 major pathways between the basal ganglia and the motor cortex that are damaged during Huntington’s disease. The indirect pathway is damaged first and then later the direct pathway. The indirect pathway inhibits movement so damage causes movement. The indirect pathway starts with the striatum signaling the external globus pallidus. From there the subthalamic nucelii gets signaled, which stimulates the internal globus pallidus. Excitation of the internal globus pallidus decreases the inhibition on the thalamus and as a result the thalamus over stimulates the motor cortex. This over signaling to the motor cortex causes the chorea seen in most HD patients. Destruction of the direct pathway and side effects from that are seen in later stage patients. The signal again starts in the striatum, but then goes to the internal globus pallidus. This causes the internal globus pallidus to increase inhibition of the thalamus so there is decreased signaling to the motor cortex. The result of damage to this pathway is the rigidity that occurs in late stage patients. The pathways that allow the basal ganglia to communicate with the motor cortex are damaged and symptoms depend on the pathway that is damaged. Cell death first occurs in the striatum, particularly at the head of the caudate nucleus. Reduced activation of the striatum is also seen in most patients. Along with all structures of the basal ganglia, the cerebral cortex also experiences neurodegeneration. Extreme cases can show up to a 25% loss of brain tissue. http://web.stanford.edu/group/hopes/cgi-bin/hopes_test/the-basic-neurobiology-of-huntingtons-disease-text-and-audio/ Conclusion Huntington’s is a genetic brain disease that results in motor, cognitive, and psychiatric symptoms. Having more than 40 CAG repeats in a specific place on chromosome four will guarantee that someone will get Huntington’s and it is a dominant trait. Those that have Huntington’s have a mutation in the huntingtin protein that results in abnormal clumping of proteins that leads to cell death. Damage to the pathways that allow communication between the basal ganglia and the motor cortex are damaged resulting in chorea and then rigidity. All structures of the basal ganglia show neurodegeneration and this occurs before any symptoms show and then continues.