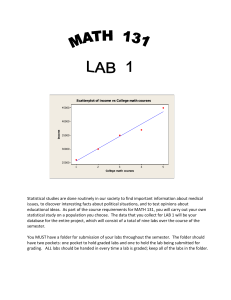
Triclinic Labs Chemometric Engine
advertisement

Triclinic Labs Chemometric Engine
Simon Bates, Ph.D.
The chemometric tools assembled at Triclinic Labs are designed to help our scientists
ask various questions about a set of measured data files:
-
How many variable components describe the differences in the observed data
sets?
How many independent solid forms (‘pure’ phases) are represented in the
data ensemble?
Clustering the data sets according to the variable components, how many of
the input data sets represent ‘pure’ phases.
How is the clustering and the relationships between clusters best displayed
visually?
With respect to the ‘pure’ phases, what is the quantitative composition of each
data set?
What do the ‘reference’ data sets corresponding to the ‘pure’ phases look
like?
What are most important observations (e.g. peaks) for classification of each
‘pure’ phase?
Are the solid-matrix effects small enough to allow traditional quantitative
analysis?
What are the magnitudes of the potential errors associated with quantitative
analysis?
How are the variable components in the data ensemble related to control
variables?
The Triclinic Labs Chemometric Engine is built around randomly generated conditional
inference trees (decision trees) [Breiman 2001 and Hothorn et al 2006]]. This allows for
an unbiased classification of the similarities and differences between a set of input data
files that is not tied to the analytical tools used to collect the data and is not dependent
on the type of material being studied. For each tree, the observations used to define the
classification of a data file at each decision node are randomly selected from the input
data set, which allows the method to identify the most predictive observations from out
of the input data set (learning algorithm). The random nature of the individual tree
growing and subsequent assembly of individual trees into multiple classification forests
further allows for an inbuilt error estimate for the classification results which is
generated as part of the classification process.
The use of conditional inference trees and random forests as a classification process is
widely used in a number of scientific disciplines, however, getting ‘appropriate’ data into
the method and interpreting the output from the method have often proved a challenge.
At Triclinic Labs we have written a number of software tools around the classification
method that take raw as measured data (structural, spectroscopic and thermal) and
convert that data into a suitable input data ensemble for classification. In each case, the
input data ensemble is representative of the measured data and does not include any
attempt to identify peaks, valleys or events of interest. The random forests method
identifies the most significant observations as part of the classification procedure in
keeping with the unbiased methodology. Similarly, the output results can be interpreted
by a number of support tools to give a meaningful overview of the classification with
respect to the questions being asked of the data by our scientists.
Because the method is a learning method and requires only the input data ensemble to
arrive at the optimum classification procedure, semi-quantitative results can be returned
without the use of any artificial standards. This is particularly useful when dealing with
production optimization or trouble shooting a production problem. For a drug product,
for example, it can be difficult (impossible) to make representative reference standards
to perform a traditional quantitative analysis. The solid state matrix and micro-structure
generated by the production process often cannot be reproduced by mixing together the
input excipients and API in known quantities. The use of conditional inference trees and
random forests can identify ‘effective reference’ patterns that correspond to the
individual phase contributions as they appear within the drug product matrix. Semiquantitative [1] analysis is performed with respect to these effective reference patterns.
[1] The method is considered to be semi-quantitative in that external reference
standards and known mixtures are not use to calibrate the output. However, in many
traditional quantitative methods, one of the largest sources of error in absolute accuracy
can be the choice of standards and the use of physical mixtures to reproduce the solidstate matrix of a drug product. This error is not identified during traditional quantitative
method development. The effective reference patterns generated by the random forests
method can be compared with the measured data collected on pure ‘standard’ material
to characterize the degree of solid-state matrix effects and its impact on absolute
accuracy. If the matrix effects are minimal then a standard quantitative analysis can be
considered to be representative of the real drug product.
For more information or to discuss how to apply the discussed techniques to your
molecules, please contact:
Simon Bates, Ph.D.
Triclinic Labs, Inc.
1201 Cumberland Ave., Suite S.
West Lafayette, IN 47906
Direct line: +1 765-588-5632
Triclinic labs: +1 765-588-6200
sbates@tricliniclabs.com
References:
Breiman L: Random forests. Machine Learning 2001; 45:5-32.
Hothorn, T., Hornik, K., & Zeileis, A. Unbiased recursive partitioning: A conditional inference
framework. Journal of Computational and Graphical Statistics 2006; 15:651– 674.
Available random forest and conditional inference tree classification software:
Breiman, L., Cutler, A., Liaw, A., & Wiener, M., 2010. randomForest: Breiman and Cutler’s
random forests for classification and regression (R package version 4.6-2) {URL:
http://cran.rproject. org/package=randomForest}
Hothorn, T., Hornik, K., Strobl, C., & Zeileis, A. (2010). PARTY: A laboratory for recursive
part(y)itioning (R package version 0.9-99991) {URL: http://cran.r-project.org/package=party}