BIOM504 - Protein sequence phylogeny
advertisement

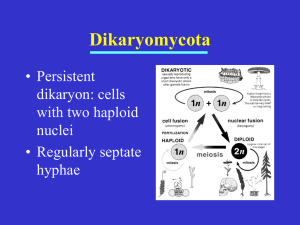
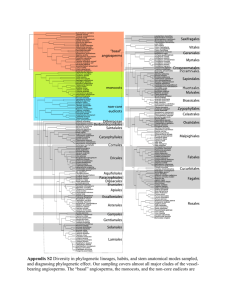
Protein sequence phylogeny Aim - to produce two phylogenetic trees using phosphoglycerate mutase sequences from a range of fungal species Files for this workshop can be downloaded from http://cogeme.ex.ac.uk/tmp/ariadne/phylogeny/ Use right click on the file to be downloaded and use ‘Save Link As’ to download the file into a directory of your choice. There are two different types of phosphoglycerate mutase (PGM). a) cofactor-dependent PGM (dPGM) uses 2,3-bisphosphoglycerate (2,3-BPG) as a cofactor. b) cofactor-independent PGM (iPGM) has two bound Mn(II) ions at its active site. Both enzymes catalyse the same overall reaction (3-phosphoglycerate → 2phosphoglycerate), but the reaction mechanisms are different and there is no sequence similarity between the enzymes. Species and sites to use for BLAST NCBI fungal genome BLAST (http://www.ncbi.nlm.nih.gov/sutils/genom_table.cgi?organism=fungi) Aspergillus nidulans FGSC A4 (filamentous ascomycete) Botryotinia fuckeliana B05.10 (filamentous ascomycete) Chaetomium globosum CBS 148.51 (filamentous ascomycete) Coccidioides immitis RS (filamentous ascomycete) Gibberella zeae PH-1 (filamentous ascomycete) Magnaporthe oryzae 70-15 (filamentous ascomycete) Neurospora crassa OR74A (filamentous ascomycete) Phaeosphaeria nodorum SN15 (filamentous ascomycete) Penicillium marneffei ATCC 18224 (filamentous ascomycete) Sclerotinia sclerotiorum 1980 UF-70 (filamentous ascomycete) Trichoderma reesei QM6a (filamentous ascomycete) Verticillium albo-atrum VaMs.102 (filamentous ascomycete) Ashbya gossypii ATCC 10895 (yeast ascomycete) Candida albicans (yeast ascomycete) Candida glabrata (yeast ascomycete) Clavispora lusitaniae ATCC 42720 (yeast ascomycete) Debaryomyces hansenii CBS767 (yeast ascomycete) Komagataella pastoris GS115 (yeast ascomycete) Kluyveromyces lactis NRRL Y-1140 (yeast ascomycete) Lodderomyces elongisporus NRRL YB-4239 (yeast ascomycete) Yarrowia lipolytica CLIB122 (yeast ascomycete) Schizosaccharomyces pombe (ascomycete, Schizosaccharomycetes) Coprinopsis cinerea okayama7#130 (basidiomycete) Cryptococcus neoformans (basidiomycete) Laccaria bicolor S238N-H82 (basidiomycete) Postia placenta Mad-698-R (basidiomycete) Puccinia graminis f. sp. tritici CRL 75-36-700-3 (basidiomycete) Schizophyllum commune H4-8 (basidiomycete) Ustilago maydis 521(basidiomycete) Encephalitozoon cuniculi GB-M1 (microsporidia) Broad Institute (http://www.broadinstitute.org/scientificcommunity/science/projects/fungal-genome-initiative/fungal-genome-initiative) Rhizopus oryzae (zygomycete) Batrachochytrium dendrobatidis (chytrid) JGI (http://genome.jgi-psf.org/) – select ‘filtered models’ Mycosphaerella graminicola (filamentous ascomycete) Phanerochaete chrysosporium (filamentous ascomycete) Phycomyces blakesleeanus (zygomycete) SGD (http://www.yeastgenome.org/cgi-bin/blast-sgd.pl) Saccharomyces cerevisiae (yeast ascomycete) Trees Create two trees, one for iPGM and one for dPGM. 1: Use the sequences below to obtain homologues of each enzyme from the following species listed above (using BLAST, e-value less than 0.0001). Ignore any hits with evalues > 1e-20. Put the sequences in a FASTA file (use Word or WordPad, but save file as a ‘Plain text file’). The names for each sequence should be 10 characters or less and only contain numbers, letters and '_'. 2: Align sequences using ClustalW (http://www.genome.jp/tools/clustalw/). Choose 'protein' and 'slow, accurate' settings. Choose 'PIR' as the output file. Save the output file to disk. 3: Use Gblocks (http://molevol.cmima.csic.es/castresana/Gblocks_server.html) to sample conserved sequence blocks from your ClustalW alignment. Use default values to start with. If Gblocks does not select any blocks then try again with "Allow gap positions within the final blocks" box ticked. Right-click on "Resulting alignment" and save to disk (in PIR format) 4: Use Readseq (http://iubio.bio.indiana.edu/cgi-bin/readseq.cgi) to change the file format of your Gblocks output from 'PIR' to 'Pearson/FASTA'. Save this file to disk. 5. Use ModelGenerator to choose the correct substitution model to use. Download ModelGenerator (file: modelgenerator.jar). Run the program using the following command: java -jar modelgenerator.jar align_file num_gamma_categories "align_file" is your output from Gblocks (in FASTA format). Set "num_gamma_categories" to 4. Look in the file “modelgenerator0.out” and the file ending in “phyml.sh” for the name of the model to use. 6: Use Readseq (http://iubio.bio.indiana.edu/cgi-bin/readseq.cgi) to change the file format of your Gblocks output from 'PIR' to 'Phylip|Phylip4'. Save this file to disk. 7: Download PhyML executable (PhyML_3.0_win32.exe) into the same folder as your alignments. PhyML will be used to create the phylogenetic tree. Type the following: PhyML_3.0_win32 -i file -d aa -b -4 -m model -v e -c 4 -a e Where “file” is the name of the Phylip formatted file created in part 6 and “model” is the substitution model from ModelGenerator. See manual (PhyML_3.0_manual.pdf ) if you are interested in what the other parameters mean. Four files will be produced, the one that ends in “tree.txt” is your tree in Newick format. Allowed Amino-acid based models: LG | WAG | JTT | MtREV | Dayhoff | DCMut | RtREV | CpREV | VT | Blosum62 | MtMam | MtArt | HIVw | HIVb | 8: View your tree using TreeDyn (http://www.phylogeny.fr/version2_cgi/one_task.cgi?task_type=treedyn), use default values. Re-root your tree using an outgroup (Homo sapiens in dGPM and E. cuniculi in iGPM). Save the final tree as a pdf. Have a look at the effect of “collapse branches with less than 50% branch support” 9: Highlight on your trees paralogous duplications (both in-paralogues and outparalogues). 10: Comment on which groups of species have each type of GPM – compare to species tree (file: 2006_Fitzpatrick_fungal_phylogeny.pdf). Sequences iPGM >XP_368343_M_oryzae MSKVEHNACLIVIDGWGVASEESPKNGDAIAAAETPVMDEFARSKTGYVELEASSLAVGLPEGLMGNSEV GHLNIGAGRVVWQDVVRIDQSIKTGEFSKNEVITQVIEAAKSGNGRLHLCGLVSHGGVAAKEAQVPKVFI HFFGDGRDTDPKSGVGYMEELLEKTKEIGIGQIATVVGRYYAMDRDKRWERVELAMKGLVLGEGEASEDP VKTVKERYEKGENDEFLKPIVVGGDEGRIKDGDNVFFFNYRSDRVRQITQLLGDVDRSPRPDFPYPKIKL ATMTRYKLDYPFDVAFEPQKMGNVLAEWLGKQNVPQVHVAETEKYAHVTFFFNGGVEKAFALEERDESQD LVPSNKSVATYDKAPEMSADGVADQVAKRLGEQKFPFVMNNFAPPDMVGHTGVYEAAIVGCAATDKAIGK IYEACKAHNYVLFITADHGNAEEMKFPDGKPKTSHTTNKVPFIMANAPEGWSLKPVGGVLGDVAPTVLAC MGLPQPEEMTALVASARVAPAPDACAPAAWFFADVERD dPGM >NP_012770_S_cerevisiae MPKLVLVRHGQSEWNEKNLFTGWVDVKLSAKGQQEAARAGELLKEKKVYPDVLYTSKLSRAIQTANIALE KADRLWIPVNRSWRLNERHYGDLQGKDKAETLKKFGEEKFNTYRRSFDVPPPPIDASSPFSQKGDERYKY VDPNVLPETESLALVIDRLLPYWQDVIAKDLLSGKTVMIAAHGNSLRGLVKHLEGISDADIAKLNIPTGI PLVFELDENLKPSKPSYYLDPEAAAAGAAAVANQGKK dPGM outgroup – add to file >AAH73741_Homo_sapiens MATHRLVMVRHGESTWNQENRFCGWFDAELSEKGTEEAKRGAKAIKDAKMEFDICYTSVLKRAIRTLWAI LDGTDQMWLPVVRTWRLNERHYGGLTGLNKAETAAKHGEEQVKIWRRSFDIPPPPMDEKHPYYNSISKER RYAGLKPGELPTCESLKDTIARALPFWNEEIVPQIKAGKRVLIAAHGNSLRGIVKHLEGMSDQAIMELNL PTGIPIVYELNKELKPTKPMQFLGDEETVRKAMEAVAAQGKAK Command prompt window Programs are run from the “Command Prompt” window (in the Start/All Programs/Accessories), used to be known as MS-DOS prompt. Useful command prompt controls: CD – change directory, can use full path (as shown in explorer window) or relative path e.g. CD C:\BLAST\data or CD data if you are already in BLAST directory. DIR – lists files in current directory The current directory appears in the command prompt window before the cursor. Phylogenetic resources Multiple sequence alignment ClustalW - http://www.genome.jp/tools/clustalw/ TCoffee - http://www.tcoffee.org/, http://www.ebi.ac.uk/Tools/t-coffee/index.html MUSCLE - http://www.drive5.com/muscle/, http://www.ebi.ac.uk/Tools/msa/muscle/ Gblocks (Selection of conserved blocks from multiple alignments) http://molevol.cmima.csic.es/castresana/Gblocks_server.html ModelGenerator (amino acid and nucleotide substitution model selection) http://bioinf.nuim.ie/modelgenerator/ Readseq (sequence format conversion tool) http://iubio.bio.indiana.edu/cgi-bin/readseq.cgi Phylogenetic analysis (mainly for download rather than online use) PhyML (A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood) - http://www.atgc-montpellier.fr/phyml/ Mr Bayes (Bayesian Inference of Phylogeny) - http://mrbayes.csit.fsu.edu/download.php PAML (Phylogenetic Analysis by Maximum Likelihood) http://abacus.gene.ucl.ac.uk/software/paml.html TREE-PUZZLE (Maximum likelihood analysis for nucleotide, amino acid, and twostate data) - http://www.tree-puzzle.de/ PHYLIP (a package of phylogenetic analysis programs) http://evolution.genetics.washington.edu/phylip.html MEGA5 (integrated phylogenetic analysis for Windows / Mac) – http://www.megasoftware.net/ Tree viewers TreeView - http://taxonomy.zoology.gla.ac.uk/rod/treeview.html TreeDyn - http://www.treedyn.org/ General http://www.phylogeny.fr/ - excellent site this, has online versions of many programs and you can join them together to do a full phylogenetic analysis.