Klekachev-SI-APL_Revision2_SRF
advertisement

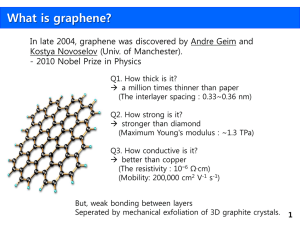
Supplementary Information Graphene as anode junction for colloidal quantum dots based light emitting diodes Alexander V. Klekachev*1,2, Sergey N. Kuznetsov3, Inge Asselberghs1,4, Mirco Cantoro1,2, Jeong Hun Mun5, Byung Jin Cho5, André L. Stesmans2, Marc M. Heyns1,6, and Stefan De Gendt1,4 1 imec, Kapeldreef 75, B-3001 Leuven, Belgium; 2 Department of Physics and Astronomy, University of Leuven, Celestijnenlaan 200D, B-3001 Leuven, Belgium 3 Department of Solid State Physics, Petrozavodsk State University, Lenin av. 33, 185910, Petrozavodsk, Russian Federation 4 Department of Chemistry, University of Leuven, Celestijnenlaan 200F, B-3001 Leuven, Belgium 5 Department of Electrical Engineering, KAIST, Daehak-Ro 291, DaeJeon, 305-701, Korea 6 Department of Metallurgy and Materials Engineering, University of Leuven, Kasteelpark Arenberg 44, B-3001 Leuven, Belgium 1. Synthesis of graphene. Characterization of graphene and ITO The chemical vapor deposition (CVD) process with inductively coupled plasma (ICP) was used for the synthesis of monolayer graphene, as it enables the growth of graphene on copper at a reduced temperature. A 300 nm thick copper layer was deposited on top of a 4-inch Si wafer with 300 nm thick SiO2 and was loaded into an ICP CVD chamber. After heating to 725°C at 50 mtorr in Ar ambient, the sample was treated with H2 plasma at a gas flow rate of 40 sccm and a RF plasma power of 50 W for 2 min. A mixture of Ar and C2H2 (Ar:C2H2 = 40:1 sccm) was then flowed into the chamber with 150 W of RF plasma for the graphene synthesis process1,2. The graphene films were transferred to 3x3 cm2 glass substrates by means of poly(methyl methacrylate) (PMMA)-supported transfer technique as described elsewhere3–5. We used a two-step procedure to strip PMMA after the transfer. First, exposure to hot acetone (50°C) for 1 h in order to remove most of PMMA. Second, another hot acetone treatment step for 15 h in order to finish the cleaning of graphene surface. This method gives an acceptable result in terms of graphene surface cleanness (Fig. S3(b)). Raman spectra of graphene films transferred to glass substrate were characterized by means of conventional confocal micro-Raman spectrometer (HORIBA Jobin Yvon LabRam 800). The excitation 532 nm diode-pumped solid state laser was focused into a spot with a diameter of 0.7 µm and safe enough power density6 of 30 kW/cm2. The spectral resolution, estimated by the full width at halfmaximum (FWHM) of one of atomic Hg emission yellow doublet lines, was 2 cm-1. A typical Raman spectrum of CVD graphene transferred onto the glass substrates is shown in Fig. S1. The narrow monotonic Lorentzian shape of 2D peak evidences the single-layer nature of graphene7. The intensity ratio of the 2D-peak to the G-peak – I2D/IG is exceeding unity and therefore, confirms the monolayer nature of the graphene and indicates a low doping level8. The D-peak is present in the spectrum, while intensity ratio ID/IG is low, indicating the low defect density and considerable crystalline quality of the graphene lattice over the whole flake area9. The obtained values are averages over 4 samples (3 points per sample area) and are presented in Table I. TABLE I. Raman spectral Raman spectra of several graphene samples Quantity ID/IG ratio I2D/IG ratio 2D-peak FWHM Value 0.18 ± 0.05 1.77 ± 0.17 38.53 ± 1.54 cm-1 Transmittance spectroscopy measurements were carried out using a two-beam visible light range spectrophotometer Shimadzu UV-1601PC. Both, indium-tin-oxide (ITO) and graphene films were measured against the bare glass substrate reference of an appropriate thickness and type taking special care of instrument’s baseline correction. Transmittance spectra of ITO and graphene are shown in Fig. S2. The average visible light range transmittance value of the different graphene samples is found to be T=97.3%, and hence, very close to the theoretical value10 of T=97.7%. Atomic force microscopy (AFM) images were taken from ITO and graphene surfaces by means of Digital Instruments Nanoscope operating in tapping mode with a scanning window size of 512x512 points. Further AFM data processing was accomplished by means of WSxM software designed by Nanotec Electronica11. Typical AFM images of ozone-treated ITO and graphene subjected to PMMA stripping procedure described above are shown in Fig. S3. The bright dots (Fig. S3(b)) are the PMMA particles that were not washed away from the graphene surface. Their appearance frequency is low as compared to the AFM images presented in other reports12. Fig. S4 shows another detailed AFM scans for both ITO and graphene samples and the respective profile trajectories. As seen from Fig. S4(c), the surface of ITO film exhibits narrow spike-like features which, according to our assumption, are responsible for higher injection capability of ITO at high current densities. 2000 Intensity (a.u.) 1800 1600 exc = 532 nm 1400 I(D)/I(G)= 0.15 I(2D)/I(G) = 1.6 1200 1000 800 600 1200 1400 1600 1800 2000 2200 2400 2600 2800 -1 Raman shift (cm ) Figure S1. Raman scattering spectrum of single layer CVD-graphene film transferred onto glass substrate. 100 Transmittance (%) 95 90 85 80 Graphene ITO 75 70 350 400 450 500 550 600 650 700 750 800 850 Wavelength (nm) Figure S2. Transmittance spectra of graphene (solid line) and ITO (dashed line) layers employed in this work. Figure S3. Atomic force microscopy images of ITO (a) and graphene (b) surfaces. The scale bar is 1 µm. (c) ITO Graphene 10 Height (nm) 8 6 4 2 0 0 200 400 600 800 Distance (nm) Figure S4. Atomic force microscopy images of ITO (a) and graphene (b) surfaces. The size of both images is 1x1 µm2. The dashed white lines show profile scan trajects with the observed profiles plotted in (c). Red and black lines in (c) correspond to ITO and graphene respectively. 2. PEDOT:PSS deposition on graphene and the effect of p-doping The deposition of poly(3,4-ethylenedioxythiophene) poly(styrenesulfonate) (PEDOT:PSS) solution on graphene surface has been studied by Park et al12 for application in graphene-based organic photovoltaic devices. Authors report on the poor wetting of graphene with PEDOT:PSS solution due to hydrophobic nature of graphene. The poor wetting of graphene surface is considered to be responsible for the low yield of working devices12. Since PEDOT:PSS is a water based solution, we performed several water contact angle (WCA) measurements on graphene and ITO for comparison purposes. WCA measurements were carried out with a computer-controlled syringe setup and an image camera for water drop image acquisition. The ITO surface has been cleaned in hot acetone followed by rinsing in isopropanol. The sample is then treated by UV-ozone for 1h under continuous O2 flow. This is a regular procedure employed for treating the ITO surface in order to improve its wettability prior to PEDOT:PSS deposition13,14. In contrast, graphene surface was not exposed to the UV-ozone as it would result in damage of graphene lattice and hence, dramatic reduce the graphene film conductivity. Water contact angles were extracted by fitting the drop contours with the elliptical or circular shapes and resulted in θ<5 and θ=63±2° for ITO and graphene respectively (Fig. S5). Authors of Ref12 demonstrate formation of non-uniform regions PEDOT layers over graphene (see Fig. S4 of supplementary information of Ref12) and they attribute it to the fact of poor wetting of graphene. However, in our experiments we were able to achieve formation of uniform and conformal PEDOT films on top of graphene. Fig. S6 shows optical photograph of typical glass substrate with graphene film on top, contacted in Van der Pauw configuration after the PEDOT:PSS deposition and baking. The sample is lying on the sheet of white paper; the image contrast is enhanced to improve the visibility of possible inhomogeneities appeared after PEDOT layer formation. However, the image reveals smooth and uniform graphene surface without evidence of any pits or islands. Moreover, our experimental results demonstrate no influence of anode type wetting properties: we obtain nearly similar yield of operating devices for both ITO and graphene based QD-LEDs. Figure S5. Images of a water drop on ITO (a) and transferred CVD-graphene on SiO2 (b). Figure S6. Contrast-enhanced optical photograph of 3x3 cm2 glass substrate (a) with a graphene flake on top covered with PEDOT. (b) Zoom-in image of (a); graphene area is outline with yellow dashed line. Note the absence of pits and islands of PEDOT over and outside the graphene. Hall-effect measurements performed before and after coating of graphene with PEDOT show strong p-doping of graphene by PEDOT. The carrier concentration increases more than four times followed by the decrease of sheet resistance after the formation of graphene-PEDOT stack. The doping level, sheet resistance and the carrier mobility before and after application of PEDOT are presented in the Table II. TABLE II. Graphene carrier concentration, sheet resistance and carrier mobility before and after PEDOT deposition as measured on one of the sample. Pristine (as prepared) After PEDOT deposition ps, 1012cm-2 3.42±0.18 13.82±0.12 Rs, Ω/☐ 1214±27 493±14 µp, cm2/Vs 1030±12 914±10 Raman spectroscopy is another powerful technique that allows us to monitor the carrier concentration in the single layer graphene films8,9,15. A shift of the 2D-peak towards higher wavenumbers is observed after the application of PEDOT on top of graphene (Figure S7). According to the Raman spectroscopy theory, this shift indicates a p-doping as compared to the pristine state. Other doping indicators such as G-peak position and I2D/IG intensity ratio are not accessible due to the overlapping of PEDOT vibrational modes16 and the stretching mode of the sp2-carbon lattice (G-peak). Pristine After PEDOT dep. 1000 Intensity (a.u.) 900 ~7cm-1 800 700 600 500 2575 2600 2625 2650 2675 2700 2725 2750 2775 -1 Raman shift (cm ) Figure S7. Raman spectra of graphene flake before and after PEDOT deposition and baking demonstrating a red-shift of the 2D band. 3. Discussion over the EL peak broadening The band broadening of the electroluminescence spectrum in the short-wavelength range, which is accompanied with a minor blue-shift of the EL peak compared to the PL spectrum peak was observed in our studies (see Fig. 1(b) of the manuscript). Fig. S8 shows the full width at half maximum (FWHM) for the EL band (left axis) and also the position of its maximum emission (right axis) for sample type 1 as a function of current density. The PL peak position and its FWHM are shown at zero current density for comparison. Note that the observed features are not unique to type 1 device, but are also pronounced for the other types of sample. This observation cannot be attributed to the quantum confined Stark effect observed in the QD layers previously17. The Stark effect would result in a clear shift of EL peak. While EL maxima has some signatures of shifting towards the short wavelength range with increase of current and/or electric field this effect is negligible and opposite in sign. In spite of that, one could clearly observe the EL band broadening towards the blue range of the optical spectrum while long-wavelength EL wing stays nearly fixed. It is known that EL peak of quantum dots has usually higher FHWM than the PL peak18, however the phenomenon presented here has never been discussed in literature. In spite of the latter, one sometimes notes a blue-tail shift when EL and PL spectra are plotted together19. Yalcin et al. recently reported on blue-shift of PL maxima from isolated negatively charged quantum dots prepared in controlled way20. The blue-shift of PL peak was attributed to the changes of electron and hole energy levels due to the excess of negative charges over the QD surface. Although authors of ref20 do not discuss the results of positive charging experiments, the spectra presented for the latter case demonstrate behavior similar to our results. This fact allows us to suppose that the increase in current density results in increasing number of (positively) charged QDs promoting the blue-shift of shortwavelength wing of the EL band. The assumption would be particularly correct if one considers the fact of charge imbalance in our structures. The observation described above needs additional experimental and theoretical investigations lying beyond the scope of this letter. 64 596 60 594 eV 2.08 2.09 592 56 2.10 590 52 588 2.11 48 EL peak position EL-peak FWHM (nm) nm 586 2.12 44 584 40 0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 2 Current density (A/cm ) Figure S8. Electroluminescence peak full width at half maximum (FWHM; open circles) and peak position (open triangles) at different excitation currents for device type 1. Values at zero current are extracted from the photoluminescence spectrum. 4. References 1 J.K. Park, S.M. Song, J.H. Mun, and B.J. Cho, Nano Letters 11, 5383 (2011). 2 T. Yoon, W.C. Shin, T.-S.Y. Kim, J.H. Mun, and B.J. Cho, Nano Letters 12, 1448 (2012). 3 S. Bae, H. Kim, Y. Lee, X. Xu, J.-S. Park, Y. Zheng, J. Balakrishnan, T. Lei, H.R. Kim, Y. Il Song, Y.J. Kim, K.S. Kim, B. Ozyilmaz, J.-H. Ahn, B.H. Hong, and S. Iijima, Nature Nanotechnology 5, 574 (2010). 4 A. Srivastava, C. Galande, L. Ci, L. Song, C. Rai, D. Jariwala, K.F. Kelly, and P.M. Ajayan, Chemistry of Materials 22, 3457 (2010). 5 X. Li, Y. Zhu, W. Cai, M. Borysiak, B. Han, D. Chen, R.D. Piner, L. Colombo, and R.S. Ruoff, Nano Letters 9, 4359 (2009). 6 B. Krauss, T. Lohmann, D.H. Chae, M. Haluska, K. von Klitzing, and J.H. Smet, Physical Review B 79, 165428 (2009). 7 A.C. Ferrari, J.C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K.S. Novoselov, S. Roth, and A.K. Geim, Physical Review Letters 97, 187401 (2006). 8 A. Das, S. Pisana, B. Chakraborty, S. Piscanec, S.K. Saha, U. V Waghmare, K.S. Novoselov, H.R. Krishnamurthy, A.K. Geim, A.C. Ferrari, and A.K. Sood, Nature Nanotechnology 3, 210 (2008). 9 A.C. Ferrari and D.M. Basko, Nature Nanotechnology 8, 235 (2013). 10 R.R. Nair, P. Blake, A.N. Grigorenko, K.S. Novoselov, T.J. Booth, T. Stauber, N.M.R. Peres, and A.K. Geim, Science 320, 1308 (2008). 11 I. Horcas, R. Fernández, J.M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero, and A.M. Baro, The Review of Scientific Instruments 78, 013705 (2007). 12 H. Park, P.R. Brown, V. Buloyic, and J. Kong, Nano Letters 12, 133 (2012). 13 K.-S. Cho, E.K. Lee, W.-J. Joo, E. Jang, T.-H. Kim, S.J. Lee, S.-J. Kwon, J.Y. Han, B.-K. Kim, B.L. Choi, and J.M. Kim, Nature Photonics 3, 341 (2009). 14 T.-H. Kim, K.-S. Cho, E.K. Lee, S.J. Lee, J. Chae, J.W. Kim, D.H. Kim, J.-Y. Kwon, G. Amaratunga, S.Y. Lee, B.L. Choi, Y. Kuk, J.M. Kim, and K. Kim, Nature Photonics 5, 176 (2011). 15 S. Pisana, M. Lazzeri, C. Casiraghi, K.S. Novoselov, A.K. Geim, A.C. Ferrari, and F. Mauri, Nature Materials 6, 198 (2007). 16 S. Garreau, G. Louarn, J.P. Buisson, G. Froyer, and S. Lefrant, Macromolecules 32, 6807 (1999). 17 S.A. Empedocles and M.G. Bawendi, Science 278, 2114 (1997). 18 H. Mattoussi, L.H. Radzilowski, B.O. Dabbousi, E.L. Thomas, M.G. Bawendi, and M.F. Rubner, Journal of Applied Physics 83, 7965 (1998). 19 L.A. Yan, J.Y. Zhang, Y.P. Cui, and Y. Qiao, Applied Physics Letters 91, (2007). 20 S.E. Yalcin, J.A. Labastide, D.L. Sowle, and M.D. Barnes, Nano Letters 11, 4425 (2011).