Word
advertisement

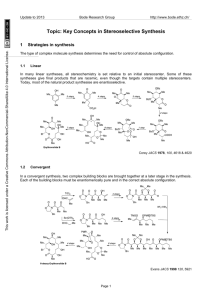
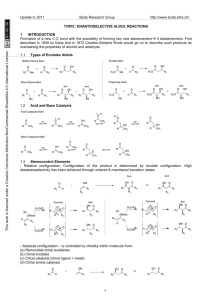
Update to 2013 Bode Research Group http://www.bode.ethz.ch/ Topic: Catalytic enantioselective conjugate additions, and cycloadditions This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. 1 Key Concepts: Regioselectivity and Catalysis 1.1 Regioselectivity 1,2- vs 1,4-Addition: α,β-unsaturated carbonyl compounds are ambident electrophiles. The incoming nucleophiles can attack either the C=O or C=C bonds O O O Nu X Nu X 1,2addition R X 1,4addition R R Nu The regioselectivity is determined by a combination of the nature of R, X and of the nucleophile: more reactive carbonyl O O Cl less reactive carbonyl + NH3 1,2-addition only NH2 O O + NH3 OMe 1,4-addition only OMe H2N Fleming, Molecular Orbitals and Organic Chemical Reactions, 2010, p 188 This selectivity can be described by the Klopman-Salem equation and Hard-Soft Acid-Base theory: Klopman, JACS, 1968, 223 HSAB theory is a useful means of classifying the reactivity of nucleophiles + electrophiles, and serves as a good “rule-of-thumb”: Pearson, JACS, 1963, 3533; Pearson, J. Chem. Ed., 1968, 581 Relevance to 1,4-conjugate addition: Properties of Acrolein Charge Distribution: Frontier Orbitals: .51 .59 LUMO -.48 Compare butadiene with acroleinO -.39 +.23 -.58 .59 HOMO -.3 O .48 LUMO coefficients correspond with increased reactivity of b-carbon -.35 -.34 -.41 Oxygen atom makes b-carbon more positive O +.48 Significantly more positive charge on C=O carbon LUMO calculations: Houk, JACS, 1973, 4094. Charges: Tidwell, JOC, 1994, 4506 These factors provide tremendous potential not only for the control of the regioselectivity, but also for the catalysis. 1.2 Common methods for achieving catalytic 1,4-conjugate additions 1 This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. Update to 2013 Bode Research Group http://www.bode.ethz.ch/ A catalyst is employed to control the regioselectivity (and ideally also the enantioselectivity) of the addition to only one of the reactive sites of an ambident molecule. 1.2.1 LUMO-Lowering by Lewis Acid Catalysis Classic Example: Diels-Alder reactions are strongly influenced by HOMO-LUMO interactions. Dramatic rate acceleration by AlCl3 catalysis observed 50 years ago: Yates, JACS, 1960, 4436 The LUMO-lowering effect of Bronsted or Lewis acids has been calculated – again on acrolein as an example: O +2.5 eV LUMO O H O H -6.5 eV O O H LUMO -14.5 eV HOMO LUMO coefficients correspond with increased reactivity of b-carbon O H -23.7 eV HOMO Houk, JACS, 1973, 4094 1.2.2 LUMO-Lowering by Catalytic Formation of Iminium Ions A more recent discovery - analogy between Lewis acid-activated unsaturated aldehydes and unsaturated iminium ions O O Lewis Acid H O R1 H R N H O R R R1 R2 • HX N R2 R1 X H (organic molecule) - H 2O LA H H (typically cationic metal) R LA N R2 H R R • Lewis acid complexation is LUMO-lowering • C=O and C=C bonds become more electrophilic • Iminium ions are positively charged • Should be more electrophilic than parent aldehyde MacMillan, JACS, 2000, 4243 1.2.3 Catalytic Generation of Soft Nucleophiles by Transmetallation: By addition of catalytic amounts of other metals, the regioselectivity of organometallic addition to unsaturated carbonyls can be altered. This was first observed in the case of Grignard reagents: Without the addition of Cu salts the 1,2-addition products (and derivatives thereof) are formed exclusively. When Cu salts are present in catalytic amounts, the 1,4-addition product is favored. HO + Et2O, 5 °C O CuCl (1 mol %) + Me Me 42% Me O Me Me MeMgBr HO Me 48% 0% Me O + + MeMgBr Et2O, 5 °C Me 7% Me Me Me 0% 1,2-Addition Products 83% Me Me 1,4-addition Kharasch, JACS, 1941, 2308 1.2.4 Catalytic Generation of Soft Anions with Chiral Bases Deprotonating weak acid with a chiral base can form a chiral ion pair that is significantly more nucleophilic than the neutral starting material: 2 This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. Update to 2013 Bode Research Group http://www.bode.ethz.ch/ Wynberg, JACS, 1981, 417 2 Catalytic, Enantioselective Conjugate Additions 2.1 LUMO lowering 2.1.1 Lewis acid activation Important Considerations Lewis acid binding to carbonyl oxygen is reversible and dynamic – for ligand and substrate design this must be taken this into account. Therefore, ligands and substrates are equipped with extra coordination sites to control these aspects to limit the number of possible transition states in the enantiodetermining step. The number of coordination sites is determined by the metal ion. Lone pair isomers + sigma bond conformers L* L* LA O C-LA sigma bond rotation L* L* LA O R2 R2 L 1* R1 R1 - anti lone pair - s-trans conformer - syn lone pair - s-trans conformer O LA L2* L2* O R2 L* O R1 L 1* R2 R1 LA LA R1 L* L* L* R2 LA O R1 - anti lone pair - s-cis conformer R2 - syn lone pair - s-cis conformer 2.1.1.1 C2-Symmetric Bis(Oxazoline) (“Box”)-Based Complexes: Cu-Bis(Oxazoline) complexes are a versatile ligand/metal combinations for a large variety of reactions (including conjugate additions and cycloadditions). The best results are obtained with substrates bearing two coordination sites to the Cu atom (they can form stable 5- or 6-membered ring chelates with the chiral Cu complex). C2-symmetry limits number of diastereomeric TS Me Me O Chiral centre from amino acids O N N Cu R TfO OTf R O Bidendate ligand for strong chelation and rigid structure, forces Cu(II) to adopt a distortet square-planar geometry RO Cu O O OR R Cu X R 6-Membered Chelate: Alkylidene Malonates 5-Membered Chelate: a-ketoesters, a-hydroxyketones ketophosphonates etc Works best on bidendate acceptors that can attach to metal with two-point binding Weakly coordinated, easily displaced counterion Evans, Acc. Chem. Res., 2000, 325 Mukaiyama-Michael addition of silyl-ketene thioacetals proceed with very high selectivity; isolation of Culigand-malonate complex showed distorted square-planar geometry; Cu-malonate in a boat conformation with shielded Re face 3 Update to 2013 Bode Research Group http://www.bode.ethz.ch/ Me Me O O Me Me N Me N Cu O MeO O H Me Me Me OMe This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. R TMSO St-Bu Me 2+ Me O O N O t-Bu O OTMS MeO N Cu 2 SbF6- t-Bu (10 mol %) OMe + HFIP, -78 °C t-BuS O R R = Ph, 91%, 93% ee R = 2-MeOPh, 92%, 99% ee R = Cy, 95%, 95% ee R = i-Pr, 93%, 93% ee CO2Me t-BuS CO2Me R Evans, JACS, 1999, 1994 6-Membered chelates based on imide frameworks places the electrophilic center roughly in the equatorial plane of the square planar complex and closer to the bulky t-Bu group of the ligand. Note that the oxazolidine moiety is installed in the substrate to provide a second coordination site for the Cu complex (It can be transformed to a variety of carboxylic acid derivatives in following steps): OTMS Top face shielded t-BuS O Me O EtO2C Me O N Cu Me O Me Me Me N - t-Bu O CO2Et O MeS OTMS Me Me O O O N Cu O N O N Me N 73%, 99:1 syn:anti, 99% ee EtO2C 2 SbF6 (10 mol %) MeS O N 2+ O N t-Bu t-BuS Me O O CO2Et O O O Me Me Me Me Bottom face exposed 90%, 5:95 syn:anti, 90% ee Evans, JACS, 2001, 4480 Activated unsaturated carbonyls that form 5-membered ring chelates have also been developed. αKetoesters are highly electron-withdrawing and make particularly reactive electrophiles: Jørgensen, ACIE, 2001, 160 Other metal-ligand complexes based on oxazoline frameworks are known. For example, Mg2+-catalyzed addition of hydrazines to imides provides pyrazolidinones: Sibi, JACS, 2007, 4522 4 Update to 2013 Bode Research Group http://www.bode.ethz.ch/ Higher oxidation state metals, for example Sc3+, can be more Lewis acidic but require an extra coordinating group to occupy 3 of 6 open sites in the octahedral geometry (in contrast to the previously mentioned square-planar geometry): Pyridine-bis(oxazoline) ligands (“PyBox”) are used: OTf O This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. O O N O X R N N N N Sc TfO OTf OTf O Sc Re face blocked N O X R Si face open Evans, JACS, 2007, 10029 This complex is an excellent catalyst for enantioselective Friedel-Crafts-type addition to unsaturated acylphosphonates- useful ester surrogates: O N + O R R O (10 mol %) Me OMe CH2Cl2, -78 °C; then DBU, MeOH O P MeO O N N N Sc TfO OTf OTf N Me R = Me, 75%, 97% ee R = Ph, 85%, 80% ee OMe Evans, JACS, 2003, 10078 2.1.1.2 Other Classes of Catalysts for Electrophile Activation: The first catalytic, enantioselective Mukaiyama-Michael reaction used a BINOL-Ti oxide complex to add to cyclic ketones: O O O O OTMS Ph + ( )n Ti O S Ph (20 mol %) PhCH3, -78 °C ( )n O Ph n = 1, 75%, 90% ee n = 2, 76%, 70% ee n = 3, 33%, 40% ee S Ph Mukaiyama, Chem. Lett. 1994, 97 Bandini and Umani-Ronchi were the first to demonstrate that one-point binding could also provide high enantioselectivity in the Friedel-Crafts alkylation: (In contrast to the previously mentioned transformations with well-defined coordination geometries). A chiral Salen ligand (Salen from salicylaldehyde and ethylenediamine) with bulky tert-butyl groups was employed, which creates a well-defined chiral geometry around the Al atom. Bandini, JOC 2004, 7517 An improved version giving better selectivities using 3,3`-disubstituted-BINOL Zirconium complexes was described (Note: The bulky 3,3’-Br substitutents on the BINOL are necessary for a high chiral induction): 5 Update to 2013 Bode Research Group http://www.bode.ethz.ch/ Br OH + Zr(OtBu)4 OH N H Br This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. R O (20 mol % each) + Ar CH2Cl2, rt O N H Indoles: 54-91%, 72-97% ee Pyrroles: 84-97%, 65-99% ee Ar R Pedro, OL, 2007, 2602 In addition to Lewis acid catalysts, Brønsted acid catalysts also promote this type of reaction: Rueping, ACIE, 2008, 593 2.1.2 Iminium catalysis - For these reaction to occur, α,β-unsaturated aldehydes (sometimes ketones) are required for the formation of the iminium ions. (This is in contrast with the metal-catalyzed reactions in Section 2.2 where unsaturated aldehydes are difficult substrates.) Therefore, the nucleophile has to be unreactive with the aldehyde (or ketone) in the absence of a catalyst to prevent an unwanted background reaction from occurring. Electrophilicity of iminiums is greater than that of aldehydes - importantly, the groups, which provide the chiral environment, are also LUMO lowering, leading to enhanced reactivity. Relative electrophilicity E of selected iminium ions O O Ph Me N Ph Ph N N N N N OTMS Me Me R LUMO lowered energy Ph Ph -10.3 -9.8 Ph Ph -8.6 Ph -8.2 -7.2 Electrophilicity E Catalytic cycle: Formation of iminium, addition of nucleophiles, followed by protonation of enamine and hydrolysis- generates and requires molecule of water for catalyst turnover Iminium catalytic cycle in nucleophilic addition to cinnamaldehyde Ph O H H Ph HNR2* · HX Ph H H2 O NR2* H X H O Nu NuH H Ph H NR2* NuH H X Mayr ACIE 2008, 47, 8723 6 Update to 2013 Bode Research Group http://www.bode.ethz.ch/ - Equilibrium between E/Z isomers leads to Curtin-Hammett control of product distribution- addition to E isomer faster than to Z Asymmetric Induction E/Z-equilibrium and preferential attack on E-isomer R' R' This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. N Nu R faster (E) R R Nu (Z) Me O N Me N Me H R1 R1 Me Me (E) x R' R' x N NH Nu Me N O slower R (Z) N x major product N N Nu E/Z :9:1 x Me O N NH x R x Me N O R1 R Nu minor product N N Nu x Nu Steric repulsion leads to high energy of both transition states MacMillan Model R1 Nu Houk Model Seebach Hev. Chim. Acta 2009, 92, 1, Hev. Chim. Acta 2009, 92, 1225, Hev. Chim. Acta 2010, 93, 1, Hev. Chim. Acta 2010, 93, 603, MacMillan JACS 2000, 122, 4243 & Houk JACS 2006, 128, 3543 - Enantioselective Friedel-Crafts alkylations MacMillan JACS 2002, 123, 4370 - Enantioselective Mukaiyama-Micheal reactions employing 2-siloxyfurans as nucleophiles to furnish butenolide structures. Here, the nature of the catalyst governs the regioselectivity: Lewis acid-based catalysts will lead to the 1,2-addition products (1,2-diol derivatives, Mukaiyama-Aldol), whereas organocatalytic approaches (iminium catalysis) will lead to the corresponding 1,4-addition products (Mukaiyama-Michael). MacMillan JACS 2003, 125, 1192 7 Update to 2013 Bode Research Group http://www.bode.ethz.ch/ Treatment of 2-siloxyfurans with a variety of ,-unsaturated aldehydes afforded syn-butenolide adducts with high diastereoselectivity and high enantioselectivity. Significantly, the use of different cocatalyst and solvent provided the opposite diastereomer with same enantioselectivity. This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. This methodology was applied in the synthesis of spiculisporic acid and 5-epi-spiculisporic acid MacMillan JACS 2003, 125, 1192 - Enantioselective Michael reactions of ,-unsaturated ketones: Jørgensen ACIE 2003, 42, 4055 - Conjugate reduction, formally, a H- is added in a 1,4-fashion to ,-unsaturated aldehydes. To render this process stereoselective, a β,β’-disubstituted aldehyde has to be used. In the following case, the Hantzsch ester (a dihydropyridine) is employed as the hydrogen source. It gets oxidized to a pyridine, thereby formally releasing an equivalent of H2. (compare also with nature’s reducing agent NADH, which essentially works via the same principle) MacMillan JACS 2005, 127, 32 - Asymmetric counteranion directed catalysis (ACDC). In this conjugate reduction of α,β-unsaturated aldehydes, the chiral 3,3’-BINOL derived phosphoric acid serves as the counterion to the iminium ion formed in situ. Electrostatic interactions keep the phosphate in close proximity to the electrophile, so that a face selection is induced. Important to note is that the bulky 3,3’substituents on the BINOL are essential for asymmetric induction to occur. 8 This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. Update to 2013 Bode Research Group http://www.bode.ethz.ch/ List ACIE 2006, 45, 4193 ,-unsaturated ketones: Neither ACDC catalysts nor chiral imidazolidinone catalysts gave good yields or enantioselectivies in the conjugate reduction of ,-unsaturated ketones. A novel class of catalytic salts, in which both anion and cation are chiral was developed by List. List JACS 2006, 128, 13368 2.2 Catalytic Generation of Soft anions 2.2.1 Soft anions by transmetallation of hard anions A wide variety of organometallic compounds can transfer their substituents (e.g. alkyl- or aryl ligands) to other catalytically active metals. Thereby, the reactivity of these substituents to be transferred can be altered. (see also section 1.2.3. : Transmetallation of a ‘hard’ Grignard reagent RMgBr to ‘soft’ copper leads to a shift of 1,2-addition to 1,4-addition) Many different organometallic reagents are either commercially available or can be prepared readily, therefore, the general approach to use them as alkyl- or aryl-donors for conjugate addition through transmetallation is attractive. 2.2.1.1 Copper-catalyzed addition of Grignard reagents Some characteristics of Cu-catalyzed enantioselective 1,4-conjugate addition of Grignard reagents are: Grignard reagents have a relatively high reactivity. The competition of fast, uncatalyzed background 1,2-carbonyl addition and 1,4-addition. Unlike in iminium catalysis, α,β-unsaturated aldehydes cannot be employed because of their high reactivity. Ketones, esters and thioesters have to be employed. + Chiral copper complexes based on phosphine-, phosphoramidite- and NHC-ligands are well known and available + Many Grignard reagents are commercially available, and inexpensive Alexakis and Baeckvall, Chem. Rev. 2008, 8, 2796, Feringa, Chem. Rev. 2008, 8, 2824 & ACIE 2010, 40, 2486. Good results with respect to yields and enantioselectivites are obtained with Cu/ferrocenyl-ligand complexes. Important to note is the fact that both cyclic and acyclic substrates give good stereoselectivities. This catalytic system works best for linear, unfunctionalized Grignard reagents. To introduce a chiral center with a methyl group (from MeMgBr), which is the most frequent pattern in natural products, one needs to use the more reactive thioesters instead of esters. 9 This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. Update to 2013 Bode Research Group http://www.bode.ethz.ch/ - Mechanism of the enantioselective 1,4 addition of ,-unsaturated carbonyl compounds promoted by copper complexes of chiral ferrocenyl diphosphine. The reaction is believed to run via a Cu-π-complex followed by a formation of a Cu-σ-complex. The transfer of R to the α,β-unsaturated carbonyl compound was found to be the rate-determining step. Feringa JACS 2006, 128, 9103 Results with ferrocenyl-based bisphosphine ligands Thioesters employed in these reactions show a more “ketone-like” reactivity and can be easily used in followup reactions, e.g. in a direct transformation to aldehydes. The aldehydes can then be employed to construct another α,β-unsaturated thioester via Horner-Wadsworth-Emmons reaction, which allows the conjugate addition to be carried out iteratively. Up to seven chiral methyl groups have been introduced using this method in the synthesis of phthioceranic acid. 10 This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. Update to 2013 Bode Research Group http://www.bode.ethz.ch/ - Similarly good results in terms of enantioselectivities can be obtained with BINAP-derived catalysts. This catalyst system requires higher temperatures than the ferrocenyl-derived one, but has a broader nucleophile scope. In contrast to the ferrocenyl-based system, one does not need to employ a thioester to achieve good enantioselectivities with MeMgBr. 2.2.1.2 Cu-Catalyzed Conjugate Addition of C-Zn and C-Al Nucleophiles + Application to All-Carbon Quaternary Centres A recent addition to enantioselective conjugate additions is the formation of very sterically hindered, quaternary all carbon centres by asymmetric conjugate addition to β,β’-disubstituted carbonyl compounds. As these substrates are less reactive due to their steric hindrance, more reactive organometallic reagents like trialkylaluminum reagents are employed. One of the first examples used highly reactive trialkylaluminums as nucleophiles with a Cu/phosphoramidite catalyst: Me Me Ar O Chiral Carbenes O Angewandte Chemie Me P N DOI : 10.1002/ anie.200604511 Me Ar Quaternary Me A ll-Carbon Stereogenic Centers by Enantioselective CuMe Catalyzed Conj ugate A dditions Promoted by a Chiral N-H eterocyclic O R = Me, R` = Et, 97% ee Carbene* * (4 mol %) O + AlR`3 R R = Et, R` = Me, 94% ee CuTC (2 mol %) M . Kevin Brown, Tricia L . M ay, Carl A . Baxter, and A mir H . R H oveyda* = i-Bu, R` = Me, 93% ee R` Et2O, -30 °C, 18 h The design of new chiral catalysts for enantioselective synthesis of all-carbon quaternary stereogenic centers is a critical and challenging objective in modern chemistry.[1] Such catalysts must be enantio-differentiating but also especially active because the desired reactions involve additions of carbon nucleophiles to sterically congested electrophilic sites. H erein, we disclose a readily available chiral N-heterocyclic carbene (NH C) that can be used to promote the catalytic asymmetric conjugate addition (A CA ) [2] of organozinc reagents to cyclic g-keto esters [Eq. (1)] ; these transformations give rise to the enantioselective formation of all-carbon R R = (CH2)2CH=CH2, R` = Me, 95% ee Alexakis, ACIE, 2005, 1376 & Chem. Commun. 2010, 46, 7295. Dialkylzinc nucleophiles are more functional group compatible than trialkylaluminums- additions to enones with β-ester groups proceed without acyl substitution and with complete regiochemistry for the more hindered site. A chiral NHC-Ag complex was employed as catalyst in this case. O + ( )n CO2Me R2Zn NHC-Ag complex (2.5 mol %) O (CuOTf)2•PhH quaternary centers that bear a readily functiona(2.5 mol stereogenic %) [3–5] lizable carboxylic ester substituent in up to 95% ee. In nearly all of the catalytic A CA reactions (one exception), the Et -30 °C ( )n 2O, new chiral NH C (Cu complex generated COin2situ) Me delivers significantly higher efficiency and asymmetric induction than R the previously reported systems. We began our investigation by examining the ability of Cu complexes generated from the reaction of 1[5a] and 2[5c] (Scheme 1) with (CuOTf) 2·C6H 6 in promoting the A CA of M e2Z n to five- and six-membered ring g-keto esters 4 and 5 (Table 1). With cyclopentenone 4a as the substrate and in the presence of binaphthyl-based 1 and (CuOTf) 2·C6H 6 [* ] M. K. Brown, T. L. May, Dr. C. A. Baxter, Prof. A. H. Hoveyda Department of Chemistry Merkert Chemistry Center Boston College Chestnut Hill, MA 02467 (USA) Fax: (+ 1) 617-552-1442 E-mail : amir.hoveyda@bc.edu 11 [* * ] We are grateful to the NIH (GM-47480) and the NSF (CHE0213009) for financial support. We thank Professor A. Alexakis (University of Geneva) for generously sharing unpublished results n = 1, R = Me, 61%, 91% ee n = 1, R = Ph, 72%, 81% ee n = 2, R = Me, 89%, 89% ee n = 2, R = i-Pr, 57%, 73% ee n = 2, R = Ph, 82%, 93% ee NHC-Ag Complex Scheme 1. Previously reported 1 and 2 and the new chiral NHC complex 3. Hoveyda, ACIE, 2007, 1097 (2.5 mol % ), less than 10% conversion is detected after 24 h (Table 1, entry 1). When biphenyl complex 2 is used, there is only 15% conversion and 6 is formed in 30% ee (Table 1, entry 2). With six-membered ring enone 5a, Cu-based complexes derived from 1 and 2 (Table 1, entries 4–5) promote the A CA of M e2Z n more efficiently (60% and > 98% conversion) and with improved enantioselectivity (73% ee and 33% ee), but only in comparison to the reactions of cyclopentenone 4a and not at synthetically useful levels. Faced with the inferior activity and asymmetric induction provided by the Cu complexes of 1 and 2, we set out to identify a more effective chiral catalyst. This initiative led us to discover that complex 3,[6] a sulfonate-containing chiral This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. Update to 2013 Bode Research Group http://www.bode.ethz.ch/ Non-cyclic unsaturated carbonyls are less reactive than cycloalkenones, thus double activation of the alkene is common. Meldrum’s acid derivatives (2 electron withdrawing groups) have been employed as activated α,β-unsaturated carbonyl compounds for the addition of dialkylzinc reagents with a Cu/phosphoramidite complex as catalyst. Fillion, JACS, 2006, 2774 Recently Hoveyda reported asymmetric conjugate additions of aryl- and alkylaluminum reagents to ,disubstituted acyclic enones. Hoveyda ACIE, 2013, 52, 8156. 2.2.1.3 Conjugate reduction A Chiral copper-hydride catalyst is prepared in situ for the asymmetric conjugate reduction of ,unsaturated carbonyl compounds. As in the case of the organocatalytic conjugate reduction (compare section 2.1.2), “H-“ is the nucleophile that is added in a 1,4-fashion. As stoichiometric reducing agents, silanes (compounds bearing a Si-H bond) are employed. PMHS is a side-product of silicone production, thus inexpensive and widely available. Buchwald JACS 2003, 125, 11253 Lipshutz and Krause, Chem. Rev. 2008, 208, 2916 12 Update to 2013 Bode Research Group http://www.bode.ethz.ch/ 2.2.1.4 Rh-Catalyzed Asymmetric Addition of Boronic Acids and their Derivatives Hayashi and Miyaura described the Rh-catalyzed addition of aryl boronic acids to unsaturated ketones in 1998 - since that time this field has developed greatly. Even though boronic acids are not “organometallic” reagents in a strict sense, they do transmetallate. This stems from the fact that the C-B bond is polarized in a similar fashion like a C-Mg bond in a Grignard reagent. This enables the R group from a boronic acid to be transferred to a metal. Boronic acids are readily available and the reaction with Rh catalysts shows a high functional group tolerance as well as mild reaction conditions. Hayashi, JACS, 1998, 5579 Mechanistic studies revealed the importance of water to help catalyst turnover and hydroxide to facilitate transmetallation and allow lower reaction temperatures: * O Ph2P PPh2 PhB(OH)2 Rh H O O H (transmetallation at 35 °C) Rh Ph H2 O Ph2P PPh2 * This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. Lipshutz JACS 2004, 126, 8352 * Ph2P O * PPh2 Rh Ph2P PPh2 Rh O P Ph Ph Ph P O Ph (S) (key aryl Rh species) si face attack O * Ph2P PPh2 Rh O Ph Hayashi, JACS, 2002, 5052 & Chem. Rev. 2003, 103, 2829. An important discovery is that chiral dienes can be used as ligands rather than chiral phosphines- this leads to far more reactive catalysts. The reactions can be carried out at much lower reaction temperatures. Hayashi, JACS, 2003, 11509 Diene ligands can provide exquisite chemo- and enantioselectivity - additions to unsaturated aldehydes proceed by 1,4-addition, not 1,2. This is in stark contrast to a Rh/phosphine catalyst, which furnishes the 1,2additon product exclusively. 13 This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. Update to 2013 Bode Research Group http://www.bode.ethz.ch/ Carreira, JACS, 2005, 10851 & Miyaura, J. Org. Chem. 2000, 65, 4450 Other boron-based reagents can also be used. Most importantly, potassium trifluoroborate salts which are air and moisture stable and easy to prepare and handle: O Me O O OMe O Me Me Ph Ph (85%, 96% ee) (51%, 91% ee) Ph (99%, 93% ee) Me O X + RBF3K ( )n (2.2 mol %) [Rh(C2H4)2Cl]2 (1.0 mol %) KOH (2.2 mol %) O O O X Et3N, PhCH3, H2O, rt ( )n Ph R (84%, 84% ee) Ph (80%, 82% ee) Darses, OL, 2009, 3486 The use of tetraarylborate salts allows synthesis of all-carbon quaternary centers. The coordination of Ar3B to the carbonyl is proposed to assist activation of the electrophile to enable attack on the hindered β-position. One drawback of this methodology is that only one of four aryl groups can be transferred: Ar3B Me Ar4BNa via O Me (2.5 mol %) R2 MeOH, Dioxane, 60 °C R3 Me Ph Me Ph R3 94%, 91% ee Rh Cl R1 R2 O O O O R1 + Ar Rh R1 R2 O Me + O O Ar R3 Ph Ar R Me R = Me, 83%, 98% ee R = CO2Me, 80%, 98% ee Ar = 4-MeC6H4, 73%, 91% ee Ar = 3-ClC6H4, 65%, 97% ee Hayashi, JACS, 2009, 13588 Boron nucleophiles can transmetallate with other soft metals beside rhodium.1,4-Additions of aryl-boronic acids to ,-unsaturated aldehydes have been performed with a combination of Pd and amine catalysts. Ibrahem & Cordova ACIE, 2013, 52, 878. 2.2.1.5 Overview of some methods for conjugate additions 14 This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. Update to 2013 Bode Research Group X Nu = CR - allylSiR3 (Nu = allyl) - Silyl-thioketene acetals - Aryls (FriedelCrafts-type) - Carbamates = OR = NR (oxazolidinones, imides) = CO2R = COH(CH3)2 = NR (Imides) = P(O)(OMe)2 = imidazoles = CR = CR Ligand Cu BOX Mg Sc BOX PyBOX - Ti-BINOL - oxazaborilidine Al BINOL One-point binding Salen Zr BINOL One-point binding One-point binding - Hydrazines - Aryls (FriedelCrafts-type) - Silyl(thio) ketene acetals - Aryls (FriedelCrafts-type) - Aryls (FriedelCrafts-type) = CR http://www.bode.ethz.ch/ Catalyst Lewis Acid catalyzed Cu BOX Organocatalysis - Aryls (Friedel- Imidazolidinone Crafts-type) - Silylenolethers (Mukaiyama-type) - Hydride: “H-“ (conjugate red.) - Hydride: “H-“ - chiral BINOLChiral counterions (conjugate based needed for ketones reduction) phosphoric acids Organometallic nucleophiles - R-B(OH)2 Rh BINAP or chiral R = aryl diene (lower temp) =H = CR (more difficult) =H = CR = CR (ketones) = OR = H (difficult) = CR = OR = SR = CR = CR Comments 2-point binding of substrate to catalyst “ACDC” - RMgBr R = alkyl (mostly unfunctionalized) - R3Al R = alkyl (mostly unfunctionalized) - R2Zn Cu Josiphos-type or BINAP 1,2- vs 1,4addition can be done by choice of catalyst Can be done iteratively Cu Phosphoramidites, NHC Quaternary centers Cu - Hydride “H-“ (from silanes) Cu Phosphoramidites, NHC BINAP, Josiphos Quaternary centers Conjugate reduction = CR = OR = NR 2.2.2 Generation of soft anions by deprotonation 2.2.2.1 Deprotonation of terminal alkynes Terminal alkynes are relatively acidic (pKa ~ 23 in H2O) and are easily deprotonated by weak bases in the presence of π-acidic Lewis acids such as Cu. However, alkynes often act as “dummy ligands” as they are poor nucleophiles; in the first catalytic, asymmetric alkyne addition, highly reactive electrophiles were used to overcome this poor reactivity: Ph Et HN Et N OH N MeO O PPh2 H O O O R = i-Pr, 94%, 95% ee R = Cy, 81%, 94% ee R = Et, 83%, 82% ee R = Ph, 64%, 83% ee Cu(OAc)2 H2O (cat) O O R O + Ph Na-(+)-ascorbate H2O, 0 °C O R Ph Carreira, JACS, 2005, 9682 15 Update to 2013 Bode Research Group http://www.bode.ethz.ch/ Alkyne1,4-addition to less electrophilic substrates can be carried out at elevated temperatures. Unfortunately alkyne dimerization is a common side reaction, which can be avoided using very bulky and electron-rich phosphine ligands. This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. O O PAr2 O PAr2 O H O + R1 (5.5 mol %) [Rh(OAc)(C2H4)]2 (2.5 mol %) O R1 Dioxane, 80 °C R2 R2 Si(i-Pr)3 Si(i-Pr)3 R1 = Ph, R2 = Me, 99%, 91% ee R1 = 2-Furyl, R2 = Me, 99%, 93% ee R1 = Me, R2 = Me, 90%, 95% ee R1 = Me, R2 = C5H11, 90%, 97% ee t-Bu Ar = OMe t-Bu Hayashi, JACS, 2008, 1577 2.2.2.2 Deprotonation of nitroalkanes and malonates Nitroalkanes (pKa ~ 10 in H2O) are readily deprotonated by weak bases - use of a bulky chiral cation gives high enantio- and diastereoselectivity in conjugate additions. Note that the chiral catalyst has to carry bulky 3,3’-substituents in order to achieve high ees. Malonates (pKa ~ 13 in H2O) are the classic Michael donors. Br Ar O O 1 mol % cat Cs2CO3 (3 equiv) H + ( )n R NO2 PhCH3, -20 °C O O N NO2 R O Ar O Ar = NO2 NO2 Ph CO2Et NO2 O 99%, 95:5 dr, 93% ee 99%, 89:11 dr, 90% ee O NO2 Me Me 94%, 95:5 dr, 80% ee 94%, 88:12 dr, 85% ee F F Maruoka, OL, 2005, 5143 Maruoka, OL, 2005, 3195 2.3 Dual Activation of Nucleophiles and Electrophiles 2.3.1 3,3`-Disubstituted BINOL Catalysts in Conjugate Additions of Boronic Acids Extensive computational studies by Goodman suggest that BINOL-derived boronates are more Lewis acidic than the parent boronates as the twisted orientation limits B-O interactions. Subsequently, this adduct with the ketone has a lower lying LUMO and a higher energy HOMO than the adduct of the parent boronate. Therefore, this is an example of a ligand-accelerated reaction. 16 Update to 2013 Bode Research Group http://www.bode.ethz.ch/ I -1.796 eV Me MeO MeO LUMO O -2.885 LUMO B 3.333 eV 4.626 eV O O I Me Ph -6.218 eV -6.422 eV Me O B Me Ph HOMO This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. HOMO Dual activation by HOMO raising and LUMO lowering Goodman, JOC, 2008, 5078 This concept has been shown to be practical in synthetic applications: Chong has demonstrated that BINOLs catalyzed the addition of alkynylboronates to unsaturated ketones; the proposed catalytic cycle is based on ligand exchange around the boron atom: Chong, JACS, 2005, 3244 Similarly high enantioselectivities were observed for attack of alkenylboronates- a six-membered ring transition state was proposed to explain the sense of addition: I OH R4 R3 OH R4 R5 B(OMe)2 I + 4 Å MS, CH2Cl2 reflux O R1 R3 R5 R2 O R1 R2 81-96%, 94-99.5% ee Chong, JACS, 2007, 4908 2.3.2 Al-Salen Catalyzed Conjugate Additions to Unsaturated Imides Jacobsen has made extensive use of salen ligands for enantioselective conjugate reactions - in the initial report on addition of hydrazoic acid (pKa 4.72 in H 2O), high ee was obtained for addition to β-alkylsubstituted imides. This was proposed to occur through a one-point binding, Lewis acid activation: (R,R)-Salen N N Back face t-Bu hindered NCOPh Al t-Bu O O N R H + HN3 Ph O N3 O t-Bu H t-Bu t-Bu (5 mol %) PhCH3, CH2Cl2, -40 °C N3 R O O N Ph H R = alkyl, 96-99%, 95-95% ee R = Ph, 58%, 60% ee R O N Al N t-Bu O O t-Bu Front face H exposed t-Bu Jacobsen, JACS, 1999, 8959 In the conjugate addition of cyanide to imides, high enantioselectivity was obtained for β-alkyl substrates but β-aryl ones showed no reactivity. An important observation was a 2nd order rate dependence on catalyst concentration, evidence for a bimetallic catalytically active species. 17 Update to 2013 Bode Research Group O N R1 Ph H + TMSCN i-PrOH This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. http://www.bode.ethz.ch/ O (S,S)-SalenAlCl (10 mol %) PhCH3, rt in-situ HCN O CN O R N Ph H R = alkyl, 70-96%, 87-98% ee R = Ph, vinyl, no reaction [SalenAlCl] in (M)2 • 104 Jacobsen, JACS, 2003, 4443 Therefore, a new ligand was designed which contains two Al-salen groups tethered together - dramatic rate acceleration and higher ee’s were observed. Importantly, previously unreactive electrophiles could be used. O N Al t-Bu O Dimer Salen (5 mol %) O N Cl Ph O N H O Ph TMSCN, i-PrOH TBME, 50 °C O t-Bu O t-Bu ( )n t-Bu Ph t-Bu O Cl O N H Me TMSCN, i-PrOH TBME, 50 °C O CN N H O Ph 65% conv 95% ee Ph O CN 47% conv 95% ee N H Me O Al N Ph Dimer Salen (5 mol %) O O t-Bu O O N Ph O N H Dimer Salen (5 mol %) Me t-Bu TMSCN, i-PrOH TBME, 50 °C O Ph O Me N H CN 32% conv t-Bu 92% ee Formation of Salen-CN as more reactive nucleophile, and Salen-Imide as more reactive electrophile Jacobsen, ACIE, 2008, 1762 18