pola28040-sup-0001-suppinfo
advertisement

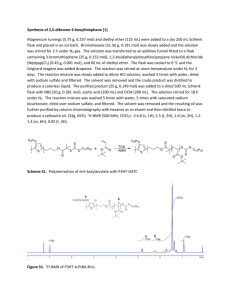
Supplementary information Poly (3-hexyl thiophene)-b-poly (N-isopropyl acrylamide): synthesis and its composition dependent structural, solubility, thermoresponsive, electrochemical and electronic properties Pallavi Kumari, Koomkoom Khawas, Sunit Hazra and Biplab Kumar Kuila*, Centre for Applied Chemistry, Central University of Jharkhand, Brambe, Ranchi, India, Pin835205 E-mail: biplab.kuila@cuj.ac.in Experimental sections Materials. All chemicals including Magnesium, 1-bromohexane, 3-bromothiophene, Na2SO4, NBromosuccinimide, Na2S2O3, KOH, Iodine, iodobenzenediacetate, 1,3-bis(diphenylphosphino) propanenickel(II) chloride [Ni(dppp)Cl2], t-BuMgCl, ethynylmagnesium bromide, N-isopropyl acrylamide, tris(2-aminoethyl)amine, formaldehyde, formic acid, CuCl, Ethyl-2-chloropropionate, DMF, NaN3 were purchased from Sigma Aldrich Pvt. Ltd. and used as received. Other solvents like THF, Ether, Methanol, Chloroform etc. were obtained from Rankem. THF was dried over Na under inert condition and used immediately for reactions. Copper (I) chloride (CuCl) was purified by washing with glacial acetic acid three times and twice with diethyl ether. N-isopropylacrylamide was recrystallized twice from benzene/hexane (60:40 v/v) prior to use. All glass apparatus for polymerization were heated and cooled down under oxygen free and moisture free argon and the reactions were performed in schlenk line. Characterization: Fourier transform infrared (FTIR) spectra in the region of 4000-500 cm-1 were recorded at room temperature using KBr pallet of the samples in a Perkin Elmer FTIR spectrometer [Spectrum two]. Nuclear magnetic resonance spectra were recorded on 200MHz Bruker instrument. Chemical shifts are expressed in parts per million (ppm) relative to the internal standard tetramethylsilane for 1H-NMR spectra. The number-average molecular weight (Mn) and polydispersity index(PDI) of P3HT samples were determined by Shimadzu prominence Gel Permeation Chromatography system in THF as solvent. The columns were calibrated against polystyrene standard samples. Atomic force microscopy (AFM) imaging was performed using AFM NT-MDT, Solver Pro 47 in the tapping mode. Analysis of the AFM images was performed with the WSxM software (Nanotec Electronica)1. Dynamic light scattering (DLS) experiments were carried out in a Malvern instrument. For temperature variable study, the solution (1 mg/ml) was heated from 250C to 450C at a rate of 20C/min and at each time after the desired temperature is reached the sample was allowed to stand at that temperature for 5 min before the measurement. The crystallographic structures of the materials were determined by a wide-angle X-ray diffraction system (Panalytical High Resolution XRD-I, PW 3040/60), Cu K-alpha radiation (40 kV, 30 mA, λ = 0.1540598 nm.Cyclic voltammetry (CV) studies were performed using a standard three-electrode system; a platinum wire was used as the counter electrode and silver wire as a pseudo reference electrode. While thin film samples solution-cast on a glassy carbon electrode were used as the working electrode, tetrabutylammoniumhexafluorophosphate (Bu4NPF6) 98% from Aldrich was used as electrolyte. All electrochemical measurements were carried out in an argon saturated electrochemical cell, which was purged with high purity argon gas for 15 min prior to each measurement. UV-vis absorption spectra were recorded in chloroform and thin film state on a Simadzu UV 3600+, UV-vis spectrophotometer. Device for mobility measurement were fabricated using ITO as an anode and Al as a cathode. In a typical experiment, poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate)(PEDOT:PSS) was spin coated on a pre-cleaned indium-tin oxide (ITO)-coated glass (∼25 Ω/sq). The ITO was cleaned first with detergent, deionizedwater, isopropyl alcohol, and acetone in an ultrasonic bath. Thin films are then spin-coated from a chlorobenzene solution (20 mg/mL) on the PEDOT:PSS layer. After thermal annealing at 140 0C for 10 min, a thin layer of Al (130 nm) was deposited on the polymer layer to complete the device fabrication. The effective area of the device is 6 mm2. The thickness of films was measured using a Daktak 6 M surface profilometer. Current voltage characteristics of the photovoltaic devices were measured with a computer-controlled Keithley 236 source measurement system. All the device manipulations and measurements were performed in inert atmosphere. Matrix assisted laser desorption ionizations spectroscopy with time of- flight detection mass spectroscopy (MALDI-TOF MS) measurements were done on a VOYAGER- DE PRO, Applied Biosystem, USA using dithranol as matrix and mixture of 1000:1 (Matrix: Polymer). Synthesis of 3-azidopropan-1-ol (1) 3-Bromo-propan -1-ol (2gm, 14.38 mmol) and sodium azide (1.62 gm, 25 mmol ) were dissolved in a mixture of acetone and water (6:1) and the resulting solution was refluxed overnight at 700C. After the completion of reaction, the acetone was removed under reduced pressure, then 30 ml of water was added and the mixture was extracted with diethyl ether. The organic layer was collected and dried over anhydrous sodium sulfate. After removal of the solvent under reduced pressure, the compound was collected as colorless oil(80%). FTIR: ̅ = (broad, OH) 3459, 2956, 2887, 2093, 1639, 1455, 1344, 1261, 1059, 955, 901, 640, 546, 513 cm -1 Synthesis of 2-bromo-2-methyl-propanoic acid-3-azido propyl ester(2) A solution of 1 (1.46 gm,) and triethylamine (2.3 ml) in 30 ml diethyl ether was cooled at 00C. 2-bromo-2-methyl propionyl bromide was added drop wise to the reaction mixture and stirred for 1 hr at 00C and left for overnight at room temperature.The white suspension reaction mixture was filtered and the filtrate pale yellow solution was washed with saturated NaHCO3solution for two times and extracted with diethyl ether. The solvent was removed under reduced pressure and the yellow oily residue was purified by silica gel chromatography, eluent as (petroliumether/Et2O, 19:1) to obtain (2) (85%).FTIR ̅ =2974, 2932, 2100, 1736, 1463, 1275, 1163, 1109 cm-1, 1 HNMR (CDCl3) (Fig. S1) Synthesis of azide functionalized PNIPAM In a typical polymerization method, 1gm (8.84 mmol) of NIPAM was taken in schlenk tube and 2-propanol (2ml) was added. The mixture was degassed by bubbling with argon for 20 min under magnetic stirring. Required amounts of Me6TREN (0.19 mmol, 46.25 mg) and initiator 2-bromo1-methyl-propinoic acid-3-azido propyl ester (0.19 mmol, 47 mg) were added under argon atmosphere. Then, CuCl (19.8 mg) was added to the reaction mixture and polymerization was initiated at room temperature for 5hrs. The reaction was ceased using liquid nitrogen and left for defreezing. The polymerization was stopped by exposing the mixture to air and added 1 to 2 mL H2O. The solvent was removed under reduced pressure and the reaction mixture wasdissolved in THF. The Reaction mixture was passed through silica gel column to remove the copper salt and after the solvent removed by rotary evaporator,residue were dissolved in THF and precipitated in excess of diethyl ether. The final product was dried in a vacuum oven at 600C, yield60%. Synthesis of alkyne terminated P3HT The synthesis of P3HT-alkyne terminated is followed from the earlier procedure2. In a Schlenk flask, 2.0 g (5.36 mmol) of 2-bromo-5-iodo-3-hexylthiophene monomer was taken into highly pure argon atmosphere and evacuated under reduced pressure to remove moisture and oxygen inside the flask. 50 milliliters of THF was added and the solution was stirred at 0 °C under argon. Equal amount of Isopropylmagnesium chloride in 2MTHF (2.68 mL, 5.36 mmol) was added drop wise and allowed to react for 30 min at 0 °C. Next, the solution was heated to 35 °C followed by the addition of Ni(dppp)Cl2(49 mg, 0.09 mmol). The resulting mixture was stirred for 30 min, cooled to 00C and further reacted with 0.5 M ethynylmagnesium bromide in THF (2.68, 1.34 mmol) for an additional 10 min. The reaction mixture was quenched in methanol to obtained dark-purple solid which was filtered and washed with excess methanol. The oligomer or low molecular weight fraction was removed by successive washing of the product with hexane and dried under vacuum to yield 0.40 g of P3HT-alk (40% yield).The same procedure was followed to synthesize a low molecular weight P3HT by varying the catalyst amount. Synthesis of block copolymer The block copolymers were synthesized by Cu catalyzed click reaction between azide terminated PNIPAM and alkyne terminated P3HT. Depending on the weight fraction of P3HT or PNIPAM in block copolymers different stoichiometric ratio of the individual homopolymers were taken for coupling reaction. In general P3HT-alkyne and PNIPAM-N3were taken in a Schlenk flask, and dissolved in dry THF under argon atmosphere. Then, CuBr and degassed PMDETA were added successively in reaction mixture and refluxed at 60 °C for24 h. After the completion of Reaction, the reaction mixture was further diluted with THF and passed through neutral alumina column to remove Cu catalyst. The solvent was removed by rotary evaporator. Depending on the composition of the block copolymer produced different purification strategy was adopted as described below. BCP2: The crude product was dissolved in DMF and filtered to remove unreacted P3HT-alkyne and the filtrate was dried by rotary evaporator to obtain the block copolymer. This process was repeated for 3 times to ensure the complete removal of unreacted P3HT-alkyne. BCP1: The P3HT-alkyne used for the preparation of the block copolymer has lower molecular weight (Mw, 1792) and is soluble in hexane. The product was dissolved in THF and precipitated in hexane and the process was repeated for three times to ensure the complete removal of the unreacted P3HT from the product. BCP3: The crude product was dissolved in THF and precipitated in excess methanol to remove the unreacted PNIPAM and the process was repeated for three times to ensure the complete removal of unreacted PNIPAM. Table-1 depicts the details of the reactants taken for synthesis of the block copolymers Sample P3HT-alkyne, PNIPAM-N3 CuBr PMDETA Yield Mn(μmole) Mn(μmole) (mmol) (mmol) BCP1 1,792(75) 4,000(37.5) 0.37 0.37 75% BCP2 10,517(17.34) 14,287 (8.67) 0.086 0.086 μmole 65% BCP3 10,517(26.25) 4,000(52.5) 0.53 0.53 60% . Figure S11H NMR spectra of 2-bromo-1-methyl-propanoic acid-3-azido propyl ester Figure S2, enlarged FTIR spectra of P3HT alkyne, PNIPAM-N3 and block copolymers in different region (a) 1000 to 700 cm-1 (b) 4000 to 3000 cm-1 (a) (a) (b) Figure S3DLS size distribution histogram of (a) BCP1 and (b) BCP 2at 250C To study the extent of end functionalization, the precursor polymer with higher molecular weight P3HT2 was analyzed by MALDI-ToF MS measurements (Fig. S4a-b) in the reflection mode and it provided proper end-group analysis. The addition of ethyne group can be seen from the major peak series belongs to P3HT + C2H + Br (e.g., for a 40-mer, 6756 g/mol = 40 × 166.3 g/mol + 25 g/mol + 79.9 g/mol) and the two minor ones to P3HT +Br + H (6732 g/mol = 40 × 166.3 g/mol + 79.9 g/mol + 1g/mol) and for P3HT + C2H + H (6678 g/mol = 40 × 166.3 g/mol + 25 g/mol + 1 g/mol). The MALDI-ToF MS results clearly indicate the successful synthesis of P3HT with sufficient end termination with ethyne group and the termination occurs at one side of P3HT. Fig.S4 (a) MALDI-ToF MS spectra measured in the reflection mode (b) magnified part in the range of 39-41 repeating units 3 Figure S5 Second heating curves from differential scanning calorimetry at a scan rate of 10 °C/min of P3HT, PNIPAMand P3HT-b-PNIPAM IV characteristics Thickness =128 nm Thickness =57 nm BCP3 Thickness =42 nm Thickness =95 nm Thickness =110 nm Figure S6 individual IV characteristics of all the samples References: 1. I. Horcas et al. Rev. Sci. Instrum. 2007, 78, 013705. 2. A. Yokoyama, R. Miyakoshi, T. Yokozawa. Macromolecules 2004, 37, 1169-1171.