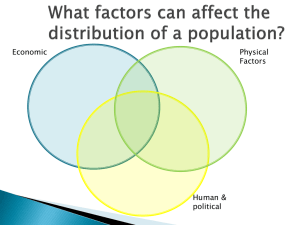
Supplementary Figure 1
advertisement

SUPPLEMENTAL DATA Supplementary Figure 1 Supplementary Figure 1: Pedigrees and Segregation of the Mutations Identified in PNPLA6. Roman numerals indicate the generation and numbers represent individuals within a generation. Circles represent females, squares represent males, filled symbols represent affected individuals, and arrows represent the proband. Abbreviations: mut – mutation and + - wild type. Supplementary Figure 2 Supplementary Figure 2: Electropherograms of Mutations Identified with Sanger Sequencing. Corresponding mutations and individuals are indicated to the left of each electropherogram. Supplementary Figure 3 Supplementary Figure 3: Structural model of EST domain (gray) of human PNPLA6. The sidechain moieties of active site residues (Ser1014/Asp1144) and those identified here to be prone to mutations (Ser1045/Thr1058/Phe1066/Val1100/Val110/Pro1122) are colored red and blue, respectively. The funnel-shaped catalytic center of the EST domain—where the active site residues Ser1014/Asp1144 are located deep at the base of the funnel while the rim of the funnel represents the entry route for the PDC substrate (indicated by the green arrow)—is represented by yellow dashed lines. Note that two different orientations of the EST domain, related by a 90-clockwise rotation about the vertical axis from left to right, are shown for the inquisitive eye. Background: PNPLA6 is constructed on the CNB1-CNB2-CNB3-EST modular architecture, where CNB1/2/3 are the N-terminal tandem cyclic nucleotide binding domains, while EST is the C-terminal phospholipid esterase domain. Notably, PNPLA6 de-esterifies phosphatidylcholine (PDC)—a major component of biological membranes—into its constituent fatty acids and glycerophosphocholine (GPC) by virtue of the esterase activity of its EST domain (5-8). The GPC product released by the catalytic action of PNPLA6 on PDC serves as a precursor for the biosynthesis of acetylcholine—a neurotransmitter involved in mediating cellular signaling in the central nervous system (CNS). The loss of catalytic function of PNPLA6 would accordingly be expected to be detrimental to the CNS homeostasis and will likely compromise the fidelity of neuronal signaling with particular impact on motor coordination. Methods: Structural models of various wildtype (WT) and mutant domains of human PNPLA6 (UniProt# Q8IY17-4) were built using the MODELLER software based on homology modeling (1). Briefly, the structural model of wildtype EST domain, which represents the site of location of Ser1045Leu, Thr1058Ile, Phe1066Ser, Val1100Gly, Val1110Met and Pro1122Leu mutations, was constructed using the crystal structure of the homologous EST catalytic domain of the plant patatin PAT17 (PDB# 1OXW) (2). A total of 100 atomic models were calculated and the structures with the lowest energies, as judged by the MODELLER Objective Function, were selected for further analysis. The atomic models were rendered using RIBBONS (4). Results:Six of the ten disease mutations identified in this study map to the EST domain of PNPLA6. Our structural model of wildtype EST domain, which represents the site of location of Ser1045Leu, Thr1058Ile, Phe1066Ser, Val1100Gly, Val1110Met and Pro1122Leu mutations, reveals that all of these six mutations will seriously jeopardize the catalytic function of PNPLA6 (Supplementary Figure 3). Notably, the EST domain of PNPLA6 is comprised of a canonical -fold shared by other members of the phospholipid esterase family (13, 14). As observed in other esterases, the catalytic center of EST domain of PNPLA6 adopts a funnel-like shape—where the active site Ser1014/Asp1144 catalytic dyad is located deep at the base of the funnel, while the rim of the funnel represents the entry route for the PDC substrate. It should be noted here that Asp1144 is believed to serve as the catalytic base that facilitates the formation of a thiolate anion on Ser1014—corresponding to the serine residue within the evolutionarily conserved GXSXG esterase motif. Subsequently, the Ser1014 thiolate anion acts as the catalytic nucleophile to initiate the cleavage of the ester bond between fatty acid chains and GPC moiety within PDC. Importantly, the location of active site residues Ser1014/Asp1144 in a deep pocket is necessary to accommodate the PDC substrate with the long fatty acid chains facing toward the rim of the catalytic funnel. In agreement with previous work conducted on esterases (15-17), the rim of the catalytic funnel of the EST domain of PNPLA6 is largely comprised of several loops that display amphipathic character. Thus, while the polar and charged residues within these loops are solvent-exposed, the hydrophobic residues point to the interior of the funnel. Accordingly—depending on the polarity of the solvent—the rim of the catalytic funnel displays a high degree of plasticity in that it can undergo constriction so as to confer a “closed” conformation which impedes the entry of the substrate, or alternatively, it can relax to give rise to an “open” conformation that is a pre-requisite for catalytic activity. Surprisingly, the Thr1058Ile/Pro1122Leu mutations map to two loops that constitute the rim of the catalytic funnel in the EST domain of PNPLA6. Our structural model suggests that the Thr1058Ile/Pro1122Leu mutations will drastically affect the amphipathic character of the catalytic funnel. Thus, for example, while Thr1058 is solvent-exposed in agreement with its polar hydroxyl sidechain, Ile1058 would be expected to point toward the interior of the funnel lined with hydrophobic residues. In particular, the newly introduced isoleucine (Ile1058) and leucine (Pro1122Leu) will likely engage via van der Waals contacts with other hydrophobic residues located within the interior of the catalytic funnel. The establishment of such new intramolecular contacts will most likely result in the spatial rearrangement of the loops resulting in the “closure” of the catalytic funnel. This will restrict the entry of PDC substrate, which in turn could dramatically affect the overall enzymatic activity of PNPLA6. On the other hand, Ser1045Leu/Phe1066Ser/Val1110Met mutations map to three neighboring -helices that together act as a scaffold for maintaining the structural integrity of the catalytic funnel. Given their strategic placement within close proximity to the catalytic center, these mutations will likely result in the structural disorganization of the catalytic funnel and misalignment of active site Ser1014/Asp1144 catalytic dyad. This in turn will likely compromise the catalytic function of PNPLA6. Finally, while the Val1100Gly mutation is located at a distal site away from the catalytic center, it may also indirectly affect protein dynamics that could ultimately shut down the enzymatic activity of PNPLA6. Supplementary Figure 4 Supplementary Figure 4: Structural models of wild type (left) and mutant (right) CNB1 (a) and CNB2 (b) domains in complex with cAMP. In each case, the CNB domain is colored yellow and cAMP red. The sidechain moieties of Val263Ile (CNB1) and Gly578Trp (CNB2) mutations identified here are depicted in blue. Background: The CNB domains are present in a wide variety of cellular proteins which act as molecular switches in response to changes in the intracellular concentration of cyclic nucleotide monophosphates (cNMPs) such as cAMP and cGMP—secondary messengers that are usually released upon the activation of transmembrane G-protein coupled receptors (GPCRs) in response to a diverse array of extracellular signals such as odors, hormones and neurotransmitters. Thus, cellular proteins harboring CNB domains conduct a plethora of physiological functions including neuronal signaling, muscle contraction and metabolic homeostasis. It is well-documented that CNB domains usually shutdown the activity of cellular proteins in which they reside in an allosteric manner and that the binding of cNMPs induces a conformational change that subsequently relieves such auto-inhibition (9-12). In light of this knowledge, it is tempting to speculate that the CNB1/2/3 domains likely play a key regulatory role in modulating the catalytic function of the EST domain of PNPLA6. More specifically, we believe that the CNB1/2/3 domains shutdown the catalytic function of the EST domain under quiescent state via an intramolecular interaction that blocks the access of PDC substrate to the enzyme active site. The binding of cNMPs to one or more CNB1/2/3 domains is likely required to rescue the enzymatic activity of PNPLA6. Methods: The structural models of various WT and mutant CNB1/2 domains in complex with cyclic adenine monophosphate (cAMP) were obtained using the crystal structure of the homologous CNB2 domain of the regulatory alpha subunit of cAMP-dependent protein kinase A bound to cAMP (PDB# 1RGS) (3). Results:The Val263Ile and Gly578Trp substitutions are respectively located within the CNB1 and CNB2 domains of PNPLA6 (Supplementary Figure 4). It is noteworthy that the CNB1/2 domains share a canonical topological fold comprised of a central -barrel supported on one end by -helices (Supplementary Figures 4a and 4b). The cAMP activator binds within the V-shaped lumen of the -barrel and it is stabilized by an extensive network of hydrophobic and electrostatic interactions. In particular, this is aided by virtue of the ability of sidechain moieties of numerous hydrophobic, polar and charged residues to protrude into the -barrel accommodating cAMP in a manner akin to that observed for the regulatory alpha subunit of cAMP-dependent protein kinase A (3). Most tellingly, our structural model of wildtype and mutant CNB1/2 domains in complex with cAMP reveal that both Val263Ile and Gly578Trp mutations occur at strategic positions within the - barrel close to the binding site of cAMP. Thus, for example, Val263 is located at the mouth of the -barrel of CNB1 domain such that the introduction of an isoleucine (Val263Ile) with a more bulkier sidechain would likely upset the balance of various hydrophobic interactions so as to destabilize the binding of cAMP (Supplementary Figure 4a). Intriguingly, the Gly578Trp mutation within CNB2 domain occurs in a structurally-equivalent position to that discussed above for the Val263Ile substitution in the CNB1 domain (Supplementary Figure 4b). However, while Val263Ile represents a relatively conservative substitution, the Gly578Trp mutation in contrast is anything but conservative. Accordingly, the effect of Gly578Trp mutation on the destabilization of -barrel of CNB2 domain would in all probability be even more dramatic than that expected for the Val263Ile mutation on the -barrel of CNB1 domain. Taken together, our structural analysis of various disease mutations identified here in PNPLA6 suggests strongly that they bear the potential to seriously affect the enzymatic activity of the EST domain via two distinct mechanisms: (i) The Ser1045Leu, Thr1058Ile, Phe1066Ser, Val1100Gly, Val1110Met and Pro1122Leu mutations will likely inactivate the EST domain by virtue of their ability to induce structural disorganization of its catalytic center; (ii) The Val263Ile and Gly578Trp mutations will likely compromise the ability of one or both regulatory CNB1/2 domains to bind cNMPs and their failure to undergo a conformational change in response to changes in intracellular concentration of cNMPs would thus keep the EST domain in an auto-inhibited state. We also note that while our structural models presented above do not account for how Gly840EGlu and Arg1362Gly mutations may compromise the cellular function of PNPLA6, both of these mutations occur in structurally flexible regions. Thus, while the Gly840Glu mutation is located in the flexible linker connecting the regulatory CNB1/2/3 domains to the catalytic EST domain, the Arg1362 is found close to the C-terminus of PNPLA6. Although it is not clear how these mutations may alter the physiological function of PNPLA6, we are tempted to speculate that the flexible regions harboring them may represent key interaction sites for as yet unidentified cellular partners of PNPLA6. Accordingly, we believe that while Gly840Glu and Arg1362Gly mutations may not directly affect the catalytic activity of PNPLA6, they could be equally detrimental in their ability to compromise its participation in other signaling cascades and/or its recruitment to specific intracellular compartments by virtue of their ability to abolish its binding to other cellular partners—that is they may be targeting its adaptor rather than enzymatic function. REFERENCES 1. Marti-Renom, M. A., Stuart, A. C., Fiser, A., Sanchez, R., Melo, F., and Sali, A. (2000) Comparative Protein Structure Modeling of Genes and Genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291-325. 2. Rydel, T. J., Williams, J. M., Krieger, E., Moshiri, F., Stallings, W. C., Brown, S. M., Pershing, J. C., Purcell, J. P., and Alibhai, M. F. (2003) The crystal structure, mutagenesis, and activity studies reveal that patatin is a lipid acyl hydrolase with a Ser-Asp catalytic dyad. Biochemistry 42, 6696-6708. 3. Su, Y., Dostmann, W. R., Herberg, F. W., Durick, K., Xuong, N. H., Ten Eyck, L., Taylor, S. S., and Varughese, K. I. (1995) Regulatory subunit of protein kinase A: structure of deletion mutant with cAMP binding domains. Science 269, 807-813. 4. Carson, M. (1991) Ribbons 2.0. J. Appl. Crystallogr. 24, 958-961. 5. Zaccheo, O., Dinsdale, D., Meacock, P. A., and Glynn, P. (2004) Neuropathy target esterase and its yeast homologue degrade phosphatidylcholine to glycerophosphocholine in living cells. J Biol Chem 279, 24024-24033. 6. Strickland, J. A., Orr, G. L., and Walsh, T. A. (1995) Inhibition of Diabrotica Larval Growth by Patatin, the Lipid Acyl Hydrolase from Potato Tubers. Plant Physiol 109, 667-674. 7. Atkins, J., Luthjens, L. H., Hom, M. L., and Glynn, P. (2002) Monomers of the catalytic domain of human neuropathy target esterase are active in the presence of phospholipid. Biochem J 361, 119-123. 8. van Tienhoven, M., Atkins, J., Li, Y., and Glynn, P. (2002) Human neuropathy target esterase catalyzes hydrolysis of membrane lipids. J Biol Chem 277, 20942-20948. 9. Kawasaki, H., Springett, G. M., Mochizuki, N., Toki, S., Nakaya, M., Matsuda, M., Housman, D. E., and Graybiel, A. M. (1998) A family of cAMP-binding proteins that directly activate Rap1. Science 282, 2275-2279. 10. de Rooij, J., Rehmann, H., van Triest, M., Cool, R. H., Wittinghofer, A., and Bos, J. L. (2000) Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J Biol Chem 275, 20829-20836. 11. VanSchouwen, B., Selvaratnam, R., Fogolari, F., and Melacini, G. (2011) Role of dynamics in the autoinhibition and activation of the exchange protein directly activated by cyclic AMP (EPAC). J Biol Chem 286, 42655-42669. 12. Rehmann, H., Das, J., Knipscheer, P., Wittinghofer, A., and Bos, J. L. (2006) Structure of the cyclic-AMP-responsive exchange factor Epac2 in its auto-inhibited state. Nature 439, 625-628. 13. Schrag, J. D., and Cygler, M. (1997) Lipases and alpha/beta hydrolase fold. Methods Enzymol 284, 85-107. 14. Kienesberger, P. C., Oberer, M., Lass, A., and Zechner, R. (2009) Mammalian patatin domain containing proteins: a family with diverse lipolytic activities involved in multiple biological functions. J Lipid Res 50 Suppl, S63-68. 15. Cygler, M., and Schrag, J. D. (1997) Structure as basis for understanding interfacial properties of lipases. Methods Enzymol 284, 3-27. 16. Schrag, J. D., Li, Y., Cygler, M., Lang, D., Burgdorf, T., Hecht, H. J., Schmid, R., Schomburg, D., Rydel, T. J., Oliver, J. D., Strickland, L. C., Dunaway, C. M., Larson, S. B., Day, J., and McPherson, A. (1997) The open conformation of a Pseudomonas lipase. Structure 5, 187-202. 17. Rubin, B. (1994) Grease pit chemistry exposed. Nat Struct Biol 1, 568-572. Supplementary Figure 5 Supplementary Figure 5: Base-pair wise coverage of PNPLA6 exons by nextgeneration sequencing reads. The y-axis depicts the coverage depth on a logarithmic scale. All bases are covered with more the 15 x and an average of 100 x, which indicates high quality data. Exons are shown in alternating colors (green and blue).