Abstract - Springer Static Content Server
advertisement

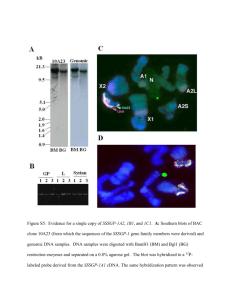
Supplemental Material A PCR Amplification Amplification of the polymorphic markers was carried out in a 25 µl-reaction containing 50 ng of DNA, 13.4 ng of each unlabeled primer, 1.5 mM dNTPs, 0.08 µg labeled primer in 1.5mM MgCl2, PCR buffer, with 1.2 U of Taq polymerase (Bio-Line, London). After an initial denaturation of 5 min at 950 C, 30 cycles were performed (940 C for 30 sec, 56-580 C for 45 sec, and 720 C for 30 sec), followed by a final extension of 10 min at 720 C. Comparative Genomic Hybridization For comparative genomic hybridization, genomic DNA was extracted from the patient and reference blood samples. The samples were labeled via nick translation, according to the manufacturer's instructions (Nick translation kit; Abbott), with the patient DNA labeled green (Green-dUTP; Abbott) and the reference DNA labeled red (Red-dUTP; Abbott). Co-precipitation of test and reference DNAs, their denaturation, along with that of the slides, and the posthybridization washes all were conducted as described previously (Fragouli et al, 2006). The hybridization was performed at 37°C for 3 days in a humid chamber, there after the slides were washed in 0.4×SSC at 75°C for 2 min and in 0.1% NP-40, 2×SSC for 2 min at room temperature. Slides were counterstained with 4,6-diamidino-2-phenylindole (DAPI) in an antifade solution. Digital Image Analysis was used to facilitate the identification of chromosomal regions with abnormal fluorescence ratios. Images of the hybridized metaphases were evaluated with a digital image analysis system based on a Zeiss fluorescence microscope and a cooled CCD camera (Photometrics, Inc., Tucson, AZ) interfaced to a Cytovision Image Analysis System (Applied 1 Imaging). Calculation of the green to red fluorescence ratios was done using Cytovision software as previously described (Israeli et al 2003). 2