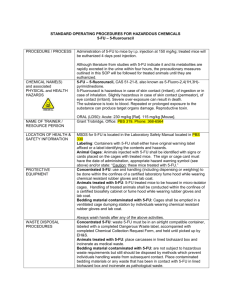
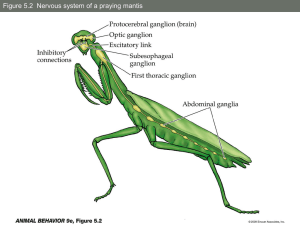
Open Access version via Utrecht University Repository
advertisement

Chronomodulated administration of Capecitabine - Aspects for clinical outcome Anna Tzani Supervisors: Prof. Dr. Jan Schellens Bart Jacobs, PharmD Institute: Division of Experimental Therapy The Netherlands Cancer Institute, Amsterdam Contents Abstract 3 1. Introduction 4 Capecitabine metabolism 4 2A. Predictive biomarkers based on enzymes expression levels 5 2A1. Predictive and prognostic value of thymidylate synthase 5 2A2. Predictive significance of thymidine phosphorylase and orotate phosphoribosyltransferase 6 2A3. Predictive importance of dihydropyrimidine dehydrogenase 6 2B. Predictive biomarkers based on enzymes genetic polymorphism 6 2C. Prominence of 5-FU/capecitabine predictive biomarkers 7 3. Circadian timing system and circadian biomarkers 8 4. The relevance of circadian timing system for anticancer treatments 10 5 The circadian variation in fluoropyrimidine enzymes 11 6 Clinical trials assessment 16 6A. Intravenous administration 16 6B. Oral administration 18 7. Comments and perspectives 21 References 25 2 Abstract The activity of the most chemotherapeutic agents is defined by the damage they can cause in healthy host tissues. This renders the establishment of the maximum tolerated dose a key step in anticancer treatment. The majority of anticancer agents, including fluoropyrimidines, exert their cytotoxicity during cell division of tumor or healthy cells. In particular, 5-fluorouracil (5-FU) and capecitabine are more toxic against cells that are in S-phase of the cell cycle. Consequently, cells that undergo DNA synthesis are more sensitive to 5-FU and capecitabine. Hence, 5-FU and capecitabine cytotoxicity depends on the cell cycle phase as well as drug metabolism and elimination. Cell division and drug exposure present circadian variation which is regulated by the circadian timing system (CTS)1. Studies in rodents have shown that several enzymes, which are important for fluoropyrimidine metabolism, show circadian variation. These enzymes include uridine phosphorylase (UP), orotate phosphoribosyl-transferase (OPRT), thymidine kinase (TK), dihydropyrimidine dehydrogenase (DPD) and thymidylate synthase (TS) that is the target enzyme of the drug2–4. Clearance of 5-FU is rate-limited by DPD. The circadian rhythm of TS and DPD has been studied in humans. Results have shown that DPD activity peaks and TS activity plunges around the middle of resting span1,5. Therefore, 5-FU and capecitabine are probably best tolerated during rest. In chronomodulated treatment, drugs are administered near their respective times of best tolerability. 5-FU and capecitabine chronotherapy is tested in several clinical trials. In some clinical trials, 5-U chronotherapy showed an improvement in tolerability versus conventional therapy. Chronomodulated treatment with capecitabine was not very advantageous in comparison to conventional treatment, for all patients. Great response variability has been observed according to gender. Men will potentially benefit from chronotherapy more than women. There is evidence that sensitivity to 5-FU and capecitabine depends on different molecular markers6. In addition, several factors like age, gender, different chronotypes and scheduling of chronotherapy can critically affect the outcome of the treatment. Therefore these factors should be taken into consideration in order to achieve more consistent results from clinical studies. 3 1. Introduction Colorectal and breast cancer remain two of the major health problems in the western world. Approximately 60% and 40% of colon and breast cancer patients respectively will develop metastatic disease and will require systemic anti-cancer therapy7,8. Fluoropyrimidines are commonly prescribed for the adjuvant treatment of epithelial cancers such as colorectal and breast tumors9. The first pyrimidine analog, 5-fluorouracil (5-FU), was synthesized more than 50 years ago and since then it has been used intravenously as chemotherapeutic regimen. Several oral prodrugs have been developed in order to overcome disadvantages from intravenous 5-FU administration. Capecitabine (N4-pentyloxycarbonyl-5deoxy-5-fluorocytidine; Xeloda, F. Hoffmann La-Roche, Basel, Switzerland) is an orally administered prodrug of 5-FU and remains the most used one among other established oral prodrugs.10 Capecitabine has been designed in a way to be mainly converted into its active form in the tumor tissue, as an attempt to avoid systemic toxicity and to increase selectivity of the drug in cancer cells 11. Tumor selectivity of capecitabine has been confirmed in animal and human studies7. Nowadays, capecitabine has replaced 5-FU in many tumor indications. It is registered for advanced colorectal cancer as monotherapy or as adjuvant treatment and for advanced breast cancer together with docetaxel or as monotherapy after failure of taxanes or anthracyclines12. Capecitabine metabolism Capecitabine is transformed into its active metabolite 5-FU by multiple sequential enzymatic reactions (fig 1). Figure 1 Simplified scheme of capecitabine metabolism and mechanism of action13. Capecitabine after absorption gets converted in three metabolic steps into 5-FU preferably in tumor tissue where the concentration of thymidine phosphorylase (TP) is higher. Further metabolism can result in two directions: elimination of the drug or activation into cytotoxic metabolites responsible for cell death. The main mechanism of action of capecitabine is inhibition of thymidylate synthase (TS) and misincorporation of cytotoxic metabolites into RNA and DNA. Abbreviations: 5′DFCR (5′-deoxy-5-fluorocytidine), 5′DFUR (5′deoxy-5-fluorouridine), 5-FU (5-fluorouracil), TP (thymidine phosphorylase), UP (uridine phosphorylase), PRPP (phosphoribosyl PPI) is a co-factor of OPRT (orotate phosphoribosyl transferase) enzyme (not shown here), DPD (dihydropyrimidine dehydrogenase), FUTP (fluorouridine triphosphate), FdUTP (fluorodeoxyuridine triphosphate) FBAL (α-fluoro-β-alanine). 4 Upon oral administration, capecitabine gets rapidly absorbed as intact molecule from the intestinal mucosa. After absorption, the molecule is metabolized by subsequently carboxylesterase (CES), cytidine deaminase (CDD) and thymidine phosphorylase (TP). These steps lead to the active form 5-FU. The third step is very important for the selectivity of the drug as TP has been found to be upregulated three to ten-fold in tumor tissues in comparison to healthy tissues10,11. Further 5-FU metabolism takes place via a number of different biochemical pathways resulting in anabolite production mainly of fluorouridine triphosphate (FUTP) and fluorodeoxyuridine triphosphate (FdUTP) which are responsible for tumor cell death. The major fraction of 5-FU is catabolised by dihydropyrimidine dehydrogenase (DPD) into biologically inactive metabolites that are excreted in the urine and the bile. This pathway represents the 80% of the drug metabolism14. The main action of the drug is the inhibition of thymidylate synthase (TS). The enzyme regulates thymidine synthesis which is essential for DNA replication. Inhibition of TS leads to apoptosis. As a consequence, the sensitivity of cancer cells to 5-FU and capecitabine depends on the extent of TS inhibition15. A different way in which the damage can occur is by misincorporation into RNA with consequent disruption of the transcription process or by misincorporation into DNA which results in DNA damage14. 2A. Predictive biomarkers based on enzymes expression levels The expression levels of TP, OPRT and DPD, which are key enzymes for capecitabine metabolism and the expression of TS, which is the main target of the drug, have been examined as possible predictive biomarkers for drug responsiveness and drug toxicity. A predictive biomarker allows the identification of the group of patients who will most likely respond to a specific therapy. Patient-tailored capecitabine therapy would potentially improve treatment efficacy and tolerability. In attempt to find acceptable biomarkers, numerous studies have focused on the relationship between TS and tumor response to Capecitabine or 5-FU treatment. Fewer studies have focused on TP, OPRT and DPD enzymes16. Most of these clinical studies were prevalently based on the expression levels of the above mentioned enzymes in tumor tissues. 2A1. Predictive and prognostic value of thymidylate synthase Jensen et al. presented a number of studies suggesting that low TS levels in tumor tissue correlate with increased progression free survival (PFS) and/or overall survival (OS) of patients treated with capecitabine and could possibly predict a better response to the drug as well16. In addition, a recent study on breast cancer patients suggested that high TS and low TP levels may be related to a reduced PFS after capecitabine treatment17. TS expression could be a promising biomarker for 5-FU and capecitabine treatment, however more analysis is required in order to reach a sufficient level of evidence18 and be recommended for routine clinical use. An obstacle in evaluation of TS levels is that they may vary between primary tumor tissue and metastasis. So, attention should be paid to the type of the tissue chosen for analysis when evaluating TS levels as a predictive biomarker19. In addition to its predictive value, TS could be also used as a prognostic marker. There are studies showing that low TS 5 levels are significantly correlated with higher overall survival20, in patients treated only with surgery (without 5-FU adjuvant treatment). 2A2. Predictive significance of thymidine phosphorylase and orotate phosphoribosyltransferase TP and OPRT enzymes are involved in capecitabine metabolism in order to form its active metabolites, as it is already discussed. Given that, one can easily conclude that high levels of these enzymes could possibly predict a better response to the drug and also higher toxicity. However, the results from the few studies available do not reveal such consistent conclusions21. Despite the important role of these two enzymes in capecitabine metabolism, they can be bypassed by other pathways involved as well in the activation of the drug. Additionally, TP is identical to platelet-derived endothelial cell growth factor. With this function the enzyme can promote angiogenesis, tumor growth and metastasis22. Therefore high TP levels in tumor of an untreated patient can confer bad prognosis, while could be possibly beneficiary in the treatment with capecitabine22. Since the role of TP and OPRT expression on 5-FU and capecitabine efficacy and toxicity is still unclear, we cannot use these markers in clinical practice yet 16. 2A3. Predictive importance of dihydropyrimidine dehydrogenase Results from studies performed to examine DPD levels in correlation to capecitabine response, suggested improved PFS and OS with low DPD levels23–25. Theoretically this is a logical conclusion as DPD activity is the rate-limiting step in catabolism and elimination of the drug. This means that low DPD levels possibly correspond to high intracellular concentrations of 5-FU and cytotoxic metabolites. On the other hand, no untreated groups were included and tumor samples were collected from primary and metastatic lesions as well. These observations lead to the hypothesis that these studies were not properly designed to completely validate DPD as predictive biomarker16. 2B. Predictive biomarkers based on enzymes genetic polymorphism A biomarker is useful not only to predict the efficacy of a certain drug but also the grade of toxicity that may be caused from this drug. In case of capecitabine, 5-FU plasma levels higher than therapeutic levels can result in increased toxicity. DPD is the rate-limiting enzyme in 5-FU catabolism and at least 80% of the drug follows this pathway. So, there is evidence that DPD deficiency will result in high 5-FU levels and increased toxicity. Loganayagam et al. reported four gene sequence variants (c.1905+1G4A, c.2846A4T, c.1601G4A and c.1679T4G) of the DPYD gene (encoding for DPD enzyme) that are associated with DPD deficiency26. In their study, all patients who were found to be heterozygous or compound heterozygous for these variants experienced severe (grade 3-4) fluoropyrimidine toxicity like diarrhea, mucositis and neutropenia26. Other studies also showed that decreased DPD enzyme activity is a significant cause for grade 3-4 toxicity27–29 providing decent evidence to sustain measurement of these variants before starting therapy. Moreover, Loganayagam et al. also agreed with other studies30–32 that there must be a 50-60% dose reduction for patients with the c.1905+1G4A variant, as this is the most 6 prominent loss-of-function allele of the DPYD gene and about 25% dose reduction for the other variants26. Among the most frequent toxicities upon capecitabine treatment are diarrhea and hand-foot syndrome. Loganayagam et al. identified two variants of the promoter region of the CDA gene (c.−92 A>G and c.−451C>T) which were related to grade 2-4 diarrhea. They have also described a homozygous MTHFR 1298CC variant genotype to be associated with hand-foot syndrome after treatment with capecitabine. However, in these cases further studies are needed in order to decide if a dose reduction strategy would be clinically advantageous26. It is true that most patients with profound DPD deficiency experience high grade of the typical 5-FU toxic effects like myelosuppression, diarrhea, mucositis and neurotoxicity33. However, high 5-FU plasma levels and consequent severe toxicity has been also seen in patients with normal DPD activity. A recent study showed that 3 out of 188 patients treated with 5-FU presented low clearance and high 5-FU plasma levels but normal DPD activity. Looking only at DPD activity, these 3 patients would not have been identified at risk to develop toxicity34. This suggests that 5-FU plasma levels resulting from capecitabine metabolism are not depending only on DPD activity and we should be very critical when a patient experiences severe toxicity. 2C. Prominence of 5-FU/capecitabine predictive biomarkers Despite the efforts to identify predictive biomarkers for capecitabine and 5-FU efficacy and toxicity, there are no consistent results suggesting one single molecule that can be extensively used as a biomarker in clinical practice, regarding the protein levels. Low levels of TS, that has been studied the most, seem to be a promising biomarker for better response to capecitabine treatment. However, there is a need for more properly designed studies to further validate this hypothesis. Regarding the association of TS levels with toxicity, results from several studies are contradictory26. It is more likely that variations in the gene encoding for TS (TYMS) resulting in higher TS protein levels could possibly be associated with adverse events, however, the effect is rather small and pre-treatment testing would not be clinically significant26,35. There is evidence that high TP/DPD enzyme ratio is associated with higher disease-free survival rate, compared to lower TP/DPD ratio in colorectal cancer patients who follow adjuvant therapy with fluoropyrimidines36. Intratumoral TP/DPD enzyme ratio could be a significant marker for adjuvant fluoropyrimidine therapy. The role of TP and OPRT levels in the evaluation of drug efficacy and toxicity is uncertain, suggesting that looking for the expression levels of only one enzyme in such a complicated metabolic pathway might be too simple16. In conclusion, many genetic polymorphisms have been identified to be related to toxicity. Therefore, upfront genotyping for the DPYD 1905+1G4A mutation can be suggested for patients treated with capecitabine. In the following paragraphs of this review we focus on a different phenomenon related to capecitabine efficacy and toxicity: the role of circadian rhythms within the capecitabine pathway. 7 3. Circadian timing system and circadian biomarkers Chronopharmacology studies the interaction of biological rhythms with the different medications. The tolerability and the antitumor effect of capecitabine highly depend on the activity of the enzymes involved in its metabolic pathway. The enzymatic activity, in many cases, presents circadian variation. Every cell has an endogenous 24h molecular clock regulated by 15 clock genes37. Cell molecular clocks constitute the circadian timing system (CTS) which drives 24h changes in xenobiotic metabolism and detoxification, cell cycle, DNA repair, apoptosis and angiogenesis38. Chronotherapy is aiming to adjust the administration of the drugs to the CTS in order to achieve better tolerability and/or efficacy of the medication. The CTS is synchronized by environmental parameters such as day-night alternation, everyday life routines and meal times. Examples of circadian rhythms in humans are the sleep-wake cycle, the core body temperature, the heart rate and the metabolism that fluctuate during a 24h period. In humans, motor activity is usually higher during day and lower during night. The maximum of body temperature is near early evening, cortisol secretion reaches the maximum near 8:00h, while melatonin secretion peaks around 2:00h. Human circadian rhythms are coordinated by the suprachiasmatic nucleus (SCN) which is a pacemaker located in the hypothalamus (fig 2)37. The SCN is regulated by daily environmental changes and produces rhythmic physiological output signals like rest-activity, hormonal secretions and body temperature. The CTS comprises the central pacemaker (SCN) and the peripheral cellular clock regulated from the 15 clock genes. Figure 2 Schematic view of the circadian mammalian system and correlation with chronotherapy 37. Circadian timing system (CTS) is composed of the suprachiasmatic nuclei (SCN) in the hypothalamus, a set of SCN-originated rhythmic physiological outputs and the molecular clock of each cell in all peripheral tissues. The CTS is synchronized by light-dark alternation and other daily behavioral activities and environmental factors. Subsequently SCN coordinates and synchronizes the peripheral molecular clocks through output signals like hormonal secretion, body temperature and rest-activity rhythm. These circadian outputs can be monitored and measured and serve as biomarkers of the CTS. Chronotherapy consist of the adaptation of drug administration to CTS in order to increase tolerability and efficacy of the drug. 8 Healthy individuals can present variations in their CTS due to differences in living habits, physiology and clock gene expression39. CTS can also be shifted, lengthened or shortened by deficiencies in recognition of external synchronizers (e.g. through blindness). Moreover, circadian and cell cycle disruption is a hallmark of cancer especially in rapidly growing tumors and advanced stage malignancies38. In turn, changes in secretion of chronobiotic agents like melatonin and glucocorticoids are happening in order to restore circadian system. Changes in rest-activity, body temperature and hormonal secretion can be used as CTS biomarkers that help in defining the circadian pattern38. When rest-activity, body temperature, plasma cortisol and melatonin concentrations reveal altered CTS activity, a personalized anticancer chronotherapy ought to be determined. Chronopharmacology investigates the dependency of the CTS on drug pharmacokinetics (PK), pharmacodynamics (PD) and the PK-PD relationship. Optimal time for drug administration, when medication will be possibly less toxic and more effective, can be predicted based on the CTS. Chronotherapy aims to adapt drug delivery schemes to circadian rhythm in order to optimize treatment effects37. 9 4. The relevance of circadian timing system for anticancer treatments The CTS rhythmically controls drug metabolism (phase I and II) and detoxification and also regulates the cell cycle of healthy cells40. CTS can modify the interactions between drugs and their molecular targets, DNA repair as well as apoptosis during the 24h period (fig 3). Clock-controlled cell proliferation determines cytotoxicity of anticancer agents as most anticancer drugs target a certain stage of cell division cycle41. Figure 3 The CTS determines the most advantageous circadian timing of anticancer treatment38. The CTS regulates transportation, bioactivation, metabolism, elimination and molecular targets that account for the chronopharmacology of anticancer drugs. The CTS also controls cell cycle events as well as DNA repair and apoptosis which elucidate the chronopharmacodynamics of anticancer agents. Therapy for which the drug administration schedule is adapted to chronopharmacokinetics and chronopharmacodynamics is named chronotherapy. It has been demonstrated in experimental systems that both dose and circadian timing play a crucial role on tumor growth inhibition and patient survival for at least 28 anticancer drugs, among which is the 5-FU42. The chronoefficacy was determined based on the administration of the drug, both as single agent and in combination with up to four other drugs at certain circadian times42. Chronoefficacy usually coincides with chronotolerance for 28 drugs tested and in general for cytostatics, interferons, antiangiogenic agents, and cell cycle inhibitors38. Hence, experimental chronotherapeutics sustains circadian timing as a possible method to enhance anticancer treatment. Chronoefficacy can arise from the circadian control of cell cycle phase related to protein targets, like TS for 5-FU. It can also derive from the circadian regulation of other enzymes involved in drug metabolism. Both concepts are illustrated in figure 3. 10 Both 5-FU and capecitabine target cells on S-phase. The main target of capecitabine and 5-FU is thymidylate synthase (TS) which presents circadian rhythm. Studies in mice have shown that the peak of bone marrow TS activity is near the second half of darkness and coincides with the highest hematologic toxicity of the drug1. Transcription and activity of TS are highest in early S-phase in proliferating tissues43. Meanwhile, bone marrow cells of mice that are in S phase increase by almost 50% in the mid dark44. This means that the circadian control of cell cycle in correlation with TS activity can affect 5-FU chronoefficacy. Other enzymes of the fluoropyrimidine metabolic pathway that present circadian organization are DPD, UP, OPRT and TK3,4. However, for some of them, their potency in the metabolism and their exact circadian pattern in humans remain to be clarified. It is also unclear whether CTS directly regulates the activity of these enzymes and it will be discussed below. 5. The circadian variation in fluoropyrimidine enzymes Cellular toxicity caused by 5-FU mainly derives from thymidylate synthase (TS) inhibition. TS activity is higher in proliferating cells than in non-proliferating45. Generally, TS activity is related to the proliferative activity of the tissue and it has been associated to higher S-phase fraction and the relative sensitivity of the tissue to 5-FU2,46. Both animal and human studies have shown that toxicity and efficacy of 5-FU depend on the time of the day at which the drug is administered47. The differences in toxicity and efficacy of the drug are possibly related to circadian organization of TS throughout the day. A recent study investigated TS activity in mice normal tissues like bone marrow, intestinal mucosa and oral mucosa, which the 5-FU damages the most2. Normal tissue damage limits 5-FU dosage. Consequently, it is possible to achieve a higher dosage when the drug is administered at the time of the highest tolerability. In this study, TS activity was monitored throughout a full 24h circadian cycle (fig 4). TS activity was found to vary almost 10-fold among different tissues and 2-fold throughout the day for the same tissue. Even though the value of their TS activity varied a lot, bone marrow and oral mucosa presented similar TS circadian rhythm with two peaks in late activity (23:00 and 21:00 hours after light onset (HALO) respectively) and in late sleep (09:30 and 09:00 HALO respectively). TS circadian rhythm in mice intestinal mucosa was slightly different with one main peak in mid-activity phase (18:45 HALO) and a smaller peak in mid-sleep phase (07:00 HALO)2. Due to circadian variability of TS, the incidence of gastrointestinal toxicity in mice can be up to 2-fold higher when capecitabine is administered at the time of lowest TS activity in comparison to administration at the time of highest TS activity. To extrapolate these results in humans we must keep in mind that humans are more active during the day while mice are more active during the night. Most physiological rhythms present a 12h phase shift between humans and rodents48. DNA synthesis in human bone marrow, oral and intestinal mucosa is almost 50% lower in the night (from 00:00 to 04:00h) in comparison to daytime49. This circadian organization of the dominant molecular target of 5-FU can partly explain why the toxic-therapeutic ratio of the drug differs within the day50. There are studies in humans that further sustain this conclusion, suggesting that CTS directly regulates the time of the day with the highest and lowest TS activity and subsequently 5-FU toxictherapeutic index1. It has been found that the protein formed from the clock gene BMAL-1 coordinates TS activity and DNA replication. High tumor nuclear BMAL-1 protein coincides with low tumor TS activity 11 and it was found to be early in the morning. Inversely, low nuclear BMAL-1 protein coincides with high tumor TS activity (during late activity)1. Low TS activity in a tissue is associated with higher toxicity in that tissue and higher tumor shrinkage upon 5-FU administration, while high TS activity is related with poor tumor response1. Figure 4 Representation of TS circadian rhythm in three different normal tissues of mice. In all three tissues we observe two peaks of TS activity during the 24h cycle and almost 2-fold differences from the highest to the lowest value within the same tissue2. Besides the importance of the circadian organization of TS, circadian activity of other enzymes within the 5-FU pathway may contribute to the toxic-therapeutic ratio of the drug. Important anabolic enzymes for fluoropyrimidine drugs like uridine phosphorylase (UP), orotate phosphoribosyl transferase (OPRT) 3 and thymidine kinase (TK)4 present circadian rhythm. Moreover, the activity of the dominant fluoropyrimidine catabolic enzyme, dihydropyrimidine dehydrogenase (DPD), is rhythmically organized throughout the day4,51. Naguib et al. (1993), described the 24h activity of hepatic OPRT and UP in female mice kept in standardized conditions of 12h light alternating with 12h darkness3. They observed a 24h cycle change in 12 the activity of the enzymes with peaks at 18 and 15 HALO and troughs at 6 and 3 HALO for OPRT and UP respectively (fig 5). Figure 5 Pattern of circadian rhythm of hepatic orotate phosphoribosyl transferase (OPRT) and uridine phosphorylase (UP) in female mice maintained under a cycle of 12h light (06:00-18:00h) and 12h darkness (18:00-06:00h)3. Rats treated with fluoropyrimidines usually experience toxicity in bone marrow, intestinal mucosa, liver and spleen4. Zhang et al. demonstrated the circadian pattern of the anabolic enzyme TK in these tissues of rats after treatment with fluoropyrimides4. The same study also showed a circadian rhythm of 5-FU catabolizing enzyme DPD activity. Moreover, they presented the relationship between the circadian organization of anabolic and catabolic enzymes activity. The animals maintained under standard light conditions from 06:00 to 18:00 and darkness from 18:00 to 06:00. The peak of TK activity in bone marrow and intestinal mucosa was at 22 HALO (04:00) and the trough at 10 HALO (16:00). TK activity, observed in the liver and spleen of the rats, peaked at 20 HALO (02:00) and showed lowest activity at 8 HALO (14:00) (fig 6). Circadian alterations of TK activity can have a great influence on bone marrow and intestinal toxicity as these are the major toxicity sites of fluoropyrimidine treatment. 13 Figure 6 Circadian pattern of TK activity in intestinal mucosa (A), bone marrow (B), liver (C) and spleen (D) of rats kept under a cycle of 12h light (06:00-18:00h) and 12h darkness (18:00-06:00h)4. The four different types of tissue present similar TK circadian rhythm. Using the same animals under the same conditions, DPD activity was measured in liver and bone marrow as well. The peak DPD activity was found to be at approximately 7 HALO (13:00) and 5 HALO (11:00) and the trough at approximately 19 HALO (01:00) and 17 HALO (23:00) for liver and bone marrow respectively (fig 7). The results revealed comparable circadian organization for TK and DPD in different tissues examined. However, a comparison of TK with DPD activity demonstrated an inverse correlation between them (when TK is at its maximum activity, DPD is reaching its minimum activity). Figure 7 Circadian pattern of DPD activity in liver (A) and bone marrow (B) of rats kept under a cycle of 12h light (06:00-18:00h) and 12h darkness (18:00-06:00h)4. The enzyme activity in the liver was shown to follow similar circadian pattern as bone marrow activity. 14 Extrapolation of circadian organization from rodents to human beings is usually difficult due to different life cycles. Rodents are more active in darkness (nocturnal) while humans are more active in the light (diurnal). Moreover, life style among humans is different because of different work schedules and other daily habits, while experimental animals under the strictly light-dark control tend to present similar circadian pattern between them. However, results from a study performed to determine DPD activity in human peripheral blood mononuclear cells5 agreed with the circadian pattern of DPD activity in rats meaning that the peak DPD activity in rodents and humans occurred at the midle stage of resting span. In this study, there was an obvious interpatient variability and the data was normalized in order to conclude to an overall pattern for all patients. The peak of DPD activity was estimated at around 01:00h and the trough at around 13:00h (fig 8). Figure 8 Circadian pattern of DPD activity in peripheral blood mononuclear cells (PBMCs) of a patient receiving continuous infusion of 5-fluorouracil5. A direct connection between DPD activity and the CTS in humans has been proved by W. Krugluger et al. They suggested that DPD transcription is controlled by the clock gene Per 1 as they noticed high correlation between DPD mRNA and Per 1 mRNA levels in colon cancer tissue52. Summarizing, the 5-FU anabolic enzymes UP, OPRT and TK, as well as the main drug-target TS and the 5FU catabolizing enzyme DPD, present circadian variations. In rodents, all these enzymes are more active during activity phase except DPD which is more active during resting phase. Obviously, since when DPD activity is low the anabolic enzymes activity is high, there may be a higher fraction of the drug entering the anabolic pathway at that time. Therefore, the exposure of cellular targets to the toxic 5-FU metabolites is lower during night, when 5-FU catabolism is accelerated. This is in agreement with the observation that 5-FU is more toxic to healthy tissues during activity span53 when DNA synthesis is higher, in both animal and humans38,54,55. In humans, it has been shown that DPD enzyme activity peaks in the middle of resting phase4,5, while the circadian organization of the anabolic enzymes UP, OPRT and TK has not been studied. So, for the human situation, data about circadian organization of 5-FU anabolic 15 enzymes is still lacking. However, regardless the circadian pattern, these anabolic pathways can be bypassed by another pathway driven by TP enzyme. TP is not circadian but it possibly drives a more dominant pathway as TP possesses high enzymatic activity, much higher than UP56. Moreover, a recent study in xenografts revealed that among fluoropyrimidine metabolic enzymes, the efficacy of capecitabine mainly depends on TP and DPD tumor levels6. Additionally, as it has been discussed, there is a directed TS regulation from the CTS through BMAL-1 protein1. In this case, DPD and TS circadian organization seem to be more important in the determination of the clinical outcome upon fluoropyrimidines administration. The abovementioned conclusions led to the hypothesis that administration of 5-FU or capecitabine at night would possibly be better tolerable from healthy tissues. Moreover, it has been noticed a correlation between 5-FU plasma levels and the efficacy of fluoropyrimidines treatment33. So, hypothetically, a higher dose administration at the time at which the drug is less toxic could achieve higher plasma levels. Several clinical trials, with 5-FU or capecitabine, have been performed in order to validate this hypothesis. 6. Clinical trials assessment The feasibility of chronotherapy with fluoropyrimidines was investigated in several clinical trials of phase I to determine safety of chronomodulation. Phase II studies were performed to determine toxicity and response of chronomodulated fluoropyrimidines. In addition, in phase III trials the chronomodulated 5FU was compared to conventional therapy. Regarding intravenous administration, different schedules were examined, where constant infusion of 5FU was mainly compared to chronomodulated infusion of 5-FU. For oral administration, different schedules were used where the daily dose was subdivided in two or three doses. A part of the dose was given during day and another part of it during evening. When assessing the applicability of chronotherapy we must keep in mind that intravenous administration has the drawback of requiring multichannel programmable pumps38, while variability in absorbance and compliance between different individuals contribute in the result of per os administration57. Chronopharmacokinetic studies are needed when drug absorbance presents high interindividual variability58. Usually, subjects participating in clinical trials tend to be compliant. However, it is useful to include assessments of compliance in the trial in order to avoid this kind of error57,59. 6A. Intravenous administration 5-FU is usually given in combination with oxaliplatin because of their synergistic effects60. However, the time of best tolerability for 5-FU is at night probably because of high DPD activity, as discussed in the previous chapter, while oxaliplatin seems best tolerable during midday61. A phase I study revealed a good safety profile for oxaliplatin with a peak delivery at 16:00h62. In a phase II trial, where patients with metastatic colorectal cancer were included, the safety and efficacy of a chronomodulated schedule were determined. The schedule consisted of oxaliplatin administrated from 10:15h till 21:45h, with a peak at 16 16:00h, combined with folinic acid and 5-FU administrated from 22:15h till 09:45h, with a peak at 04:00h. The dose of oxaliplatin, folinic acid and 5-FU were 20 mg/m²/day, 300 mg/m²/day and 600 mg/m²/day, respectively. This schedule was maintained on day 1-5 of each three-week course63. This chronomodulated combination was compared to continuous administration of the same combination of drugs in the same dosage given again for 5 consecutive days every third week as it has been investigated in two independent randomised phase III trials50,64 (table 1). In the first trial, 92 patients were included who have not previously received chemotherapy64. This study demonstrated that the chronomodulated schedule was less toxic and more effective. Patients, upon chronomodulated treatment, presented higher response rate and longer progression free survival and overall survival compared with the constant rate infusion. However, this trial was terminated prematurely because of possible chemical interactions between 5-FU and oxaliplatin when delivered simultaneously from the same catheter64. The second trial involved 186 patients with previously untreated metastases50. Half of them received chronotherapy and the other half constant rate infusion. The main clinical endpoint was the objective response rate and it was found to be higher by 21.5% in the chronotherapy group than in the constant infusion group (p = 0.003). Chronotherapy was also found to be less toxic. The incidence of severe mucosal toxicity, peripheral sensitive neuropathy, as well as other common toxic effects like diarrhea, hand-foot syndrome and neutropenia was 14% for chronotherapy vs 76% for constant infusion group (p < 0.0001). In general, lower incidence of severe toxicities in the chronotherapy arm led to less hospitalization for toxicity and lower number of patients who had to be withdrawn because of toxicity. In addition, the reduction of severe toxicities in the chronotherapy arm allowed the increase of the drug dose. This resulted in higher plasma levels and greater efficacy of the drug. In this trial, the median dose of 5-FU per course was 40% higher for chronomodulated delivery (3500 mg/m²) compared to constantrate infusion (2500 mg/m²). Another important observation was that after treatment, more patients that received chronotherapy were eligible for surgery of some metastases that were initially unresectable. Finally, the differences in median progression free survival and overall survival were not significant between the two arms, but that was not the endpoint of the trial50. Table 1 Overview of the key clinical trials investigating 5-FU administration in constant or chronomodulated schedules. 17 • Abbreviations. Ref: reference number, m/f: male/female, PFS: progression-free survival, OS: overall survival, ORR: overall response rate, chron.: chronomodulated administration, 5-FU: 5-fluorouracil, hfs: hand-foot syndrome, const.: constant infusion. In a recent meta-analysis study, data from three phase III randomised clinical studies were combined. This study revealed that only male patients benefit from chronomodulated 5-FU administration65. A total of 845 (346 females and 499 males) metastatic colorectal cancer patients were treated with chronoFLO vs conventional 5-FU schedule50,64. No significant gender differences regarding response rate (RR), progression-free survival (PFS) and overall survival (OR) were seen on the conventional therapy group. However, on the chronomodulated arm, men had significantly higher RR, PFS and OS than men in constant rate group. Contrarily, the chronomodulated administration presented similar or worse efficacy compared to the conventional infusion in women. This may happens because of different circadian genotypes between male and female patients or due to gender dependency of circadian pharmacology of anticancer drugs. The study revealed significant gender differences regarding RR (p=0.007), PFS (p=0.006) and OS (p=0.002) in response to chronotherapy and suggested that men with colorectal cancer can benefit more from chronotherapy in comparison to conventional therapy of 5-FU. Another study also showed that chronomodulated delivery of fluoropyrimidines may be beneficiary only for men but not for women66. Therefore, more attention should be paid in gender differences for future trials65. 6B. Oral administration It has been previously discussed that capecitabine is designed to be preferentially converted to 5-FU in the tumor tissue and therefore it is qualified as tumor selective11. The efficacy of capecitabine as single drug was found to be similar to 5-FU/FA (folinic acid)67 and in combination with oxaliplatin (XELOX) was found to be analogous to 5-FU/FA plus oxaliplatin (FOLFOX)68. Moreover, intravenously administrated drugs, like 5-FU, have several drawbacks such as infections, bleeding, thrombosis, pneumothorax and inconvenience for patients. Thus, capecitabine is a good alternative solution for chemotherapy. Chronomodulated administration of capecitabine was tested in several phase I and II clinical trials. The rationale to deliver a higher evening dose was driven by the circadian organization of the enzymes involved in capecitabine metabolism in addition to the preceding data regarding 5-FU. Santini et al. investigated the efficacy and toxicity of a chronomodulated schedule of capecitabine and oxaliplatin (XELOX) in advanced colorectal cancer patients resistant to 5-FU chemotherapy69. When oxaliplatin is co-administered with capecitabine, apart from the synergistic effect, it also improves tumor-selective activation of capecitabine as oxaliplatin upregulates TP expression only in tumor tissues70. In total, 36 metastatic colorectal cancer patients were included in the study. In this phase II study, oxaliplatin was continuously infused from 08:00h to 20:00h on days 1 and 8. Capecitabine was administered orally in 3 unequal doses every day: 25% of the dose at 08:00h, 25% of the dose at 18:00h and 50% of the total dose at 23:00h on day 1-14 every 3 weeks. The daily dose of oxaliplatin was 70 mg/m² and for capecitabine 1,750 mg/m². The overall response rate estimated from this study was 30.6%, while disease stabilization was achieved in 36.1% of patients. The majority of toxic effects were of grade 1 or 2, which is considered as mild or moderate. Few patients experienced grade 3 toxicities 18 including diarrhea, fatigue, mucositis, nausea, neurotoxicity and only two patients hand-foot syndrome. In general, this combined continuous infusion of oxaliplatin with chronomodulated administration of capecitabine demonstrated good overall survival and median time to progression69 (table 2). Results from this trial show that this regimen is well tolerated and presents good safety profile69. Based on the above evaluations, the group performed a phase I study with the goal to determine the maximumtolerated dose and the dose-limiting toxicities of chronomodulated capecitabine monotherapy. 27 patients with advance cancer were involved in this trial. The schedule of capecitabine administration was equal to the previous study71. They observed that the chronomodulated administration of capecitabine was safe and well tolerated in the majority of patients at the recommended dose of 2,750 mg/m/day. At this dosage the toxicities were prevalently hand-foot syndrome, fatigue and gastrointestinal toxicity. However, almost all cases were reversible after dose modification. Only one patient presented fatigue of grade 4. The tolerability of this schedule was confirmed after treatment of five cycles71. The same research group conducted another phase II study using the same drug schedule (fixed-rate infusion of oxaliplatin with chronomodulated capecitabine) including this time 46 chemonaive patients with advanced colorectal cancer72. Results from this study showed high tumor control, and overall survival, good progression-free survival and very good safety profile. It is possible that the favourable toxicity profile in these studies derives from the circadian organisation of the drug metabolism, elimination and therapeutic targets. It can also be related to the new 3-times administration schedule of capecitabine instead of the usual 2-times per day. Moreover, the 12h continuous infusion of oxaliplatin partially coincides with its optimal administration time which can also results in lower toxicity. It is particularly interesting that results were more promising than expected and consistent with other studies in untreated patients72. However, it is not safe to extract solid conclusions from these trials as there was no comparator arm. Later, Qvortrup et al. performed a phase II study with chronomodulated XELOX (XELOX30chron). The primary end point of this study was the response rate (RR) and secondary end points were toxicity, progression-free survival (PFS) and overall survival (OS)73. The trial involved 71 patients with metastatic colorectal cancer, pre-treated with 5FU and irinotecan. They designed a XELOX schedule that mimicked a chronomodulated regimen but for practical, economical reasons and for patients’ convenience the regimen was administered as follows: oxaliplatin was administered intravenously (30min infusion with dose 130 mg/m²) on the first day between 13:00 and 15:00h. Oxaliplatin induces infusion rate dependent neurotoxicity and a 2h infusion is recommended to reduce the incidence of neurotoxicity. However, in a pilot study where oxaliplatin was delivered as 30 min infusion, it has not been observed higher neurotoxicity74. A total daily dose of 2000 mg/m² of capecitabine was given twice per day in two unequal doses. 20% of the dose was given in the morning (between 07:00 and 09:00h) and 80% was delivered in the evening (between 18:00 and 20:00h). The main efficacy and toxicity results from XELOX30chron can be seen in table 2. These were similar to what Santini et al. found for their chronomodulated XELOX schedule used as second line therapy69 apart from neurotoxicity which was lower for XELOX30chron. 19 The safety (mostly regarding the grade 3 toxicity) and efficacy of XELOX30chron were also comparable with a previous study conducted by the same group, using this time a conventional schedule of capecitabine in combination with oxaliplatin75. Again, in this phase II trial, 71 pre-treated patients with advanced colorectal cancer were involved. Equal doses of capecitabine (1000 mg/m²) were administered twice per day with an interval of 12h, from day 1 to day 14. Oxaliplatin was given as 30min infusion in a dose of 130 mg/m²/day. A multicenter phase II study was conducted comparing XELOX30 and XELOX30chron with the primary objective of reducing toxicity76. The advantage of this study was that the results of the two different therapeutic schedules could be compared directly. In this trial, 141 patients with metastatic unresectable and untreated (except from previous 5-FU or capecitabine treatment ended 6 months before the study) adenocarcinoma of colon or rectum were enrolled. Patients were separated in two arms and randomly received XELOX30 (arm A) or XELOX30chron (arm B) as first line therapy. Both arms received 30min infusion of 130 mg/m² oxaliplatin on the first day of each cycle. However, in arm A the infusion time was between 08:30h and 16:00h but it was not systematically registered, while in arm B oxaliplatin was given between 13:00h and 15:00h. A total daily dose of 2000 mg/m² of capecitabine was delivered for 14 days to each group. In arm A, patients were treated with conventional capecitabine schedule receiving half of the daily capecitabine dose in the morning and half in the evening. Patients in arm B received 20% of the dose (400 mg/m²) in the morning between 07:00h and 09:00h, and 80% of the dose (1600 mg/m²) in the evening between 18:00h and 20:00h. Treatment was repeated every third week. The primary endpoint was to determine whether toxicity was reduced. The reduction of toxicity from arm A to arm B was only 5% (p=0.47), as toxic effects > grade 2 were seen in 85% of the chronomodulated arm and 90% of the conventional capecitabine therapy. Mild neurotoxicity was 15% for the conventional therapy and 13% for the chronomodulated scheme. However, the incidence of severe neurotoxicity was higher in group B that received chronotherapy. Furthermore, no significant differences were found between arm A and B, regarding median progression-free survival (8.9 vs 8.8 months) and overall survival (17.6 vs 15.5 months)76. Thus, in this study, the chronomodulated XELOX did not seem to reduce toxicity in comparison to the conventional XELOX schedule. On the other hand, the chronomodulated administration of 5-FU with oxaliplatin reduced toxicity compared with fixed infusion, according to previous studies50,64. Table 2 summarizes the main findings of studies with chronomodulated capecitabine. In general, it has been observed that patients receiving more than six cycles of chronomodulated XELOX tend to develop less severe neurotoxicity in comparison with other XELOX regimens73. Overall, the trials described, using the oral chronomodulated administration of capecitabine, were not able to demonstrate a clear time-dependency of safety and efficacy of the drug. 20 Table 2 Overview of the key clinical trials investigating the chronomodulated or conventional capecitabine administration. • Abbreviations. Ref: reference number, m/f: male/female, PFS: progression-free survival, OS: overall survival, ORR: overall response rate, chron.: chronomodulated administration, 5-FU: 5-fluorouracil, hfs: hand-foot syndrome, const.: constant infusion. 7. Comments and perspectives The circadian rhythm of TS and DPD has been studied in rodents and humans as well. According to these findings, it has been hypothesized that treatment with fluoropyrimidines would probably be best tolerated during resting phase. Based on that, chronomodulated intravenous administration of 5-FU and oral administration of capecitabine have been both tested in several clinical trials. Great response variability according to gender has been observed. Men will potentially benefit more from chronotherapy compared to women. Moreover, results from 5-FU chronotherapy seem to be more promising than capecitabine chronotherapy. This may have several explanations and one may be that antitumor activity of 5-FU and capecitabine depends on different molecular markers. According to a recent study performed in xenografts, there is great evidence that the sensitivity to capecitabine depends more on mRNA expression levels of TP, TS and the ratio of TP to DPD, while 5-FU sensitivity depends more on OPRT, UP and CDD expression6. This leads to the hypothesis that among fluoropyrimidine metabolic enzymes that present circadian organization, only DPD is really important for capecitabine sensitivity, while 5-FU sensitivity depends more on the circadian metabolic enzymes. However, further investigation is needed in order to confirm these findings in humans, since the abovementioned study was performed in xenograft models. Moreover, the circadian pattern of OPRT and UP has not been studied in humans. 21 An additional reason for which capecitabine chronotherapy did not show a clear time-dependency could be the design of the trials regarding the kind of patients selected, the dosage and the administration of the drug. Besides, capecitabine chronomodulated administration may not mimic 5-FU chronomodulated administration because of extra metabolic steps required for capecitabine bioactivation77,78 and absorption process58. Two very important factors for the trial design are patient selection and the determination of information deriving from each patient group. There are well documented differences in drug metabolism between males and females regarding 5-FU, resulting in very different toxic pattern79. Females presented much higher rate of severe toxic effects than males during treatment with chronomodulated 5-FU/FA and oxaliplatin80 which may be explained by the lower 5-FU clearance in females compared to males81. Moreover, the female circadian structure is somehow different from the male one, presenting higher melatonin rhythm suppression by light at night82 and higher cortisol response to stress83. However, all the above mentioned studies did not examine toxicity pattern according to gender. The age is another variable that can influence the absorption, the metabolism as well as the elimination of capecitabine and 5-FU. It has been recently reported higher capecitabine Cmax and AUC in patients over 70 than under 60 years which may alter the pattern of toxicity84. Hepatic and renal dysfunctions are a common problem in elderly and can affect both drug metabolism and elimination. Blood flow is very important for drug transportation. According to the size and the metabolic function each organ receives a certain fraction from the cardiac output. The higher the amount of blood that an organ receives the higher the drug rate that can transfer to the extravascular sites where the pharmacological receptors are. Blood flow and cardiac output decline with age by almost 1% every year after the age of 25 years85. These and other phenomena of age, like several possible comorbidities and the concurrent uptake of other medications, can modify the metabolism of capecitabine/5-FU as well as the circadian pattern of the patient. Another drawback of the above mentioned clinical trials is that the age range of the enrolled patients was very wide (ranging on average from 28 to 76 years old). This means that, it is very difficult to draw safe results for efficacy/toxicity of the drug from such a group. Inter-individual variations of circadian rhythms can affect drug exposure as well. There are people that demonstrate opposite circadian patterns and present higher drug (5-FU) concentration in the evening than in the morning86. Dose and time of drug administration are also important determinants of the outcome of the trials especially the chronomodulated ones. The recommended dose for capecitabine is 2500 mg/m²/day orally given twice per day usually at the end of the meal. The proposed duration is 14 days with 1 week rest period in 3-week cycles70. However, capecitabine trials that have been discussed used lower dose than the recommended one and in none of these trials the dose exceeded 2000 mg/m²/day. The time of capecitabine delivery may be crucial in a chronomodulated study. It has been reported that 5-FU concentration reaches its peak about 2h after oral administration of capecitabine70 and besides that the optimal time for 5-FU is at 04:00h. Therefore, early evening oral administration (18:00-20:00h) which has been reported in most of the studies, will result in the higher concentration of the drug much earlier than the optimal time. Furthermore, as it is already discussed, the infusion of oxaliplatin was not accomplished at the same time for both arms. 22 The biggest difference between 5-FU and capecitabine chronomodulated administration is that 5-FU is delivered intravenously while capecitabine is given orally. Oral drugs undergo an absorption process that may exhibit circadian variation due to biological daily fluctuations58. Absorption of oral administered drugs can be affected by several factors regarding the physicochemical properties of the drug as well as patient-related factors like gastrointestinal mobility and gastric emptying, gastric pH, gastrointestinal blood flow and the activity of protein-transporters and enzymes on gut surface87. As gastric pH, emptying time and intestinal mucosa blood flow vary during the day, differences in drug absorption in humans have been reported88. Gastric pH presents circadian rhythm modified by meals and nocturnal duodenal-gastric reflux89. This can influence the ionization of drugs depending on day or night administration. In general, lipophilic drugs are absorbed more rapidly when administered in the morning compared to evening administration. This can lead to higher Cmax and shorter Vmax during morning administration in contrast to evening administration58. Capecitabine is a lipophilic drug. Given that, the dose delivered in the evening can result in very different 5-FU plasma levels compared to an equal intravenously administered 5-FU dose. The rate of drug absorption can also be influenced from the gastrointestinal perfusion as blood flow affects the passage of drugs through biological membranes, especially lipophilic drugs that do not require active transportation58. Gastrointestinal blood flow is increased in early morning78. This fact may explain a higher absorption of capecitabine in the morning than in the evening. Furthermore, hepatic perfusion is higher in early morning as well78. The bioactivation of capecitabine mainly happens in the liver11 which means that the higher the hepatic blood flow is the more drug available for bioactivation. Three enzymes are involved in the bioactivation of capecitabine: (a) carboxylesterase (CES) that is mainly expressed in the liver and in the intestine90, (b) cytidine deaminase (CDA) that can be found in many tissues including liver and tumor tissues and (c) thymidine phosphorylase (TP) which is also expressed in many tissues and can be found in higher levels in some tumor tissues compared to the normal ones90. It is not precisely known if there is no circadian variation in any of these three enzymes. The pharmacokinetic parameters of capecitabine and its metabolites, 5'-deoxy-5-fluorocytidine (5'DFCR), 5'-deoxy-5-fluorouridine (5'-DFUR) and 5-FU, present high inter-individual variability as also happens with other cytotoxic drugs70. In a recently performed study, it has been reported that coefficients of variance of the concentration in plasma of capecitabine and its metabolites ranged between 36% and 142%91. Researchers believe that this is most likely because of variability of expression and activity of enzymes involved in capecitabine bioactivation91. Mercier et al described a case of CDA overexpression (180% higher activity compared to normal population) that led to unexpected and severe toxicities because of extensive activation of capecitabine and overexposure to 5-FU92. High inter-patient variability was also observed in TP expression levels in various tissues including liver and tumor tissues93. This may also influence capecitabine pharmacokinetics. Recent pharmacokinetic guided studies revealed that, among other factors, dose adjustment based on pharmacokinetic monitoring may reduce the toxicity and increase the efficacy of capecitabine. Gamelin et al performed a randomized phase III study where they noticed that pharmacokinetically guided 5-FU dose adjustment increased the response and decreased the incidence and severity of toxicity94. Moreover, Saif et al, after reviewing the various strategies developed to enhance 5-FU clinical activity, 23 mentioned that pharmacokinetically guided dose adjustments can improve the therapeutic index of 5FU33. Pharmacokinetics of capecitabine and its metabolites can potentially be useful for monitoring and guiding the therapy with capecitabine or 5-FU. Nowadays, there are methods for the quantitative determination of capecitabine and its metabolites which can be very helpful clinical studies95. It cannot be excluded that a schedule of chronomodulated administration is possibly able to increase efficacy and reduce toxicity of capecitabine. However, there is a need for further investigation of capecitabine chronopharmacokinetics and better design of trials in order to validate if there is timedependency of capecitabine therapeutic index. Based on the above-mentioned observations and the findings from the trials discussed, several adjustments can be done in order to receive more clear information from future trials. It would be safer to compare chronomodulated to conventional administration in untreated patients as their circadian pattern is not disrupted by previous anticancer medications. Results should be compared separately in men and women as gender differences influence both efficacy and toxicity of the drug. Safer conclusions would be drawn from a less wide age range of enrolled patients. Moreover, we observed that three times delivery of capecitabine was less toxic from that of two times per day. So, it would be better to use this schedule in future studies. The optimal time for drug delivery can also be improved. Based on circadian DPD activity, the best tolerability of 5-FU is near the peak of enzyme activity, around 01:00h, that has not been tested so far. However, there are big circadian differences among individuals according to gender, age, polymorphisms and chronotype41. So, to take full advantage of cancer chronotherapy and draw safe conclusions from prospective studies, there is a need for patient-tailored chronotherapy. This can be done with the help of mathematical models that take into consideration many parameters like circadian physiology, genetic background, gender, age etc. Finally, the large progress in drug-development technologies can facilitate personalized chronotherapy schedules. 24 References 1. Wood, P. A., Du-Quiton, J., You, S. & Hrushesky, W. J. M. Circadian clock coordinates cancer cell cycle progression, thymidylate synthase, and 5-fluorouracil therapeutic index. Molecular cancer therapeutics 5, 2023–33 (2006). 2. Lincoln, D. W., Hrushesky, W. J. & Wood, P. A. Circadian organization of thymidylate synthase activity in normal tissues: a possible basis for 5-fluorouracil chronotherapeutic advantage. International journal of cancer. Journal international du cancer 88, 479–85 (2000). 3. Naguib, F. N., Soong, S. J. & El Kouni, M. H. Circadian rhythm of orotate phosphoribosyltransferase, pyrimidine nucleoside phosphorylases and dihydrouracil dehydrogenase in mouse liver. Possible relevance to chemotherapy with 5-fluoropyrimidines. Biochemical pharmacology 45, 667–73 (1993). 4. Zhang, R., Lu, Z., Liu, T., Soong, S. J. & Diasio, R. B. Relationship between circadian-dependent toxicity of 5-fluorodeoxyuridine and circadian rhythms of pyrimidine enzymes: possible relevance to fluoropyrimidine chemotherapy. Cancer research 53, 2816–22 (1993). 5. Harris, B. E., Song, R., Soong, S. J. & Diasio, R. B. Relationship between dihydropyrimidine dehydrogenase activity and plasma 5-fluorouracil levels with evidence for circadian variation of enzyme activity and plasma drug levels in cancer patients receiving 5-fluorouracil by protracted continuous infusion. Cancer research 50, 197–201 (1990). 6. Yasuno, H. et al. Predictive markers of capecitabine sensitivity identified from the expression profile of pyrimidine nucleoside-metabolizing enzymes. Oncology Reports 29, 451–458 (2013). 7. Vincenzi B, Cesa AL, Santini D, Schiavon G, Grilli C, Graziano F, T. G. Predictive factors for response to chemotherapy in colorectal cancer patients. Crit Rev Oncol Hematol. 45–60 (2004). 8. Bonotto, M. et al. Making Capecitabine Targeted Therapy for Breast Cancer: Which is the Role of Thymidine Phosphorylase? Clinical breast cancer 1–6 (2012).doi:10.1016/j.clbc.2012.10.002 9. Saif, M. W. An adverse interaction between warfarin and fluoropyrimidines revisited. Clinical colorectal cancer 5, 175–80 (2005). 10. Aprile, G., Mazzer, M., Moroso, S. & Puglisi, F. Pharmacology and therapeutic efficacy of capecitabine: focus on breast and colorectal cancer. Anti-cancer drugs 20, 217–29 (2009). 11. Miwa, M. et al. Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. European journal of cancer (Oxford, England: 1990) 34, 1274–81 (1998). 12. Schellens, J. H. M. Capecitabine. The oncologist 12, 152–5 (2007). 13. Chung, Y.-L. et al. Noninvasive measurements of capecitabine metabolism in bladder tumors overexpressing thymidine phosphorylase by fluorine-19 magnetic resonance spectroscopy. 25 Clinical cancer research: an official journal of the American Association for Cancer Research 10, 3863–70 (2004). 14. Longley, D. B., Harkin, D. P. & Johnston, P. G. 5-fluorouracil: mechanisms of action and clinical strategies. Nature reviews. Cancer 3, 330–8 (2003). 15. Iqbal, S. & Lenz, H. J. Determinants of prognosis and response to therapy in colorectal cancer. Current oncology reports 3, 102–8 (2001). 16. Jensen, N. F. et al. Predictive biomarkers with potential of converting conventional chemotherapy to targeted therapy in patients with metastatic colorectal cancer. (2012). 17. Lee, S. J. et al. Thymidylate synthase and thymidine phosphorylase as predictive markers of capecitabine monotherapy in patients with anthracycline- and taxane-pretreated metastatic breast cancer. Cancer chemotherapy and pharmacology 68, 743–51 (2011). 18. Simon, R. M., Paik, S. & Hayes, D. F. Use of archived specimens in evaluation of prognostic and predictive biomarkers. Journal of the National Cancer Institute 101, 1446–52 (2009). 19. Aschele, C., Debernardis, D., Tunesi, G., Maley, F. & Sobrero, A. Thymidylate Synthase Protein Expression in Primary Colorectal Cancer Compared with the Corresponding Distant Metastases and Relationship with the Clinical Response to 5-Fluorouracil. Clin. Cancer Res. 6, 4797–4802 (2000). 20. Edler, D. et al. Thymidylate synthase expression in colorectal cancer: a prognostic and predictive marker of benefit from adjuvant fluorouracil-based chemotherapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 20, 1721–8 (2002). 21. Koopman, M., Venderbosch, S., Nagtegaal, I. D., Van Krieken, J. H. & Punt, C. J. A review on the use of molecular markers of cytotoxic therapy for colorectal cancer, what have we learned? European journal of cancer (Oxford, England: 1990) 45, 1935–49 (2009). 22. Bronckaers, A., Gago, F., Balzarini, J. & Liekens, S. The Dual Role of Thymidine Phosphorylase in Cancer Development and Chemotherapy. Med Res Rev 29, 903–953 (2009). 23. Ichikawa, W. et al. Combination of dihydropyrimidine dehydrogenase and thymidylate synthase gene expressions in primary tumors as predictive parameters for the efficacy of fluoropyrimidinebased chemotherapy for metastatic colorectal cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 9, 786–91 (2003). 24. Vallböhmer, D. et al. DPD is a molecular determinant of capecitabine efficacy in colorectal cancer. International journal of oncology 31, 413–8 (2007). 25. Salonga, D. et al. Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clinical cancer research: an official journal of the American Association for Cancer Research 6, 1322–7 (2000). 26 26. Loganayagam, A. et al. Pharmacogenetic variants in the DPYD, TYMS, CDA and MTHFR genes are clinically significant predictors of fluoropyrimidine toxicity. British journal of cancer 108, 2505–15 (2013). 27. Amstutz, U., Froehlich, T. K. & Largiadèr, C. R. Dihydropyrimidine dehydrogenase gene as a major predictor of severe 5-fluorouracil toxicity. Pharmacogenomics 12, 1321–36 (2011). 28. Loganayagam, A. et al. The contribution of deleterious DPYD gene sequence variants to fluoropyrimidine toxicity in British cancer patients. Cancer chemotherapy and pharmacology 65, 403–6 (2010). 29. Morel, A. et al. Clinical relevance of different dihydropyrimidine dehydrogenase gene single nucleotide polymorphisms on 5-fluorouracil tolerance. Molecular cancer therapeutics 5, 2895– 904 (2006). 30. Yang, C. G. et al. DPD-based adaptive dosing of 5-FU in patients with head and neck cancer: impact on treatment efficacy and toxicity. Cancer chemotherapy and pharmacology 67, 49–56 (2011). 31. Van Kuilenburg, A. B. P. et al. Evaluation of 5-fluorouracil pharmacokinetics in cancer patients with a c.1905+1G>A mutation in DPYD by means of a Bayesian limited sampling strategy. Clinical pharmacokinetics 51, 163–74 (2012). 32. Deenen, M. J. et al. Relationship between single nucleotide polymorphisms and haplotypes in DPYD and toxicity and efficacy of capecitabine in advanced colorectal cancer. Clinical cancer research: an official journal of the American Association for Cancer Research 17, 3455–68 (2011). 33. Saif, M. W., Choma, A., Salamone, S. J. & Chu, E. Pharmacokinetically guided dose adjustment of 5-fluorouracil: a rational approach to improving therapeutic outcomes. Journal of the National Cancer Institute 101, 1543–52 (2009). 34. Bocci, G. et al. A pharmacokinetic-based test to prevent severe 5-fluorouracil toxicity. Clinical pharmacology and therapeutics 80, 384–95 (2006). 35. Jennings, B. A. et al. Functional polymorphisms of folate metabolism and response to chemotherapy for colorectal cancer, a systematic review and meta-analysis. Pharmacogenetics and genomics 22, 290–304 (2012). 36. Mori, T. et al. Factors predicting the response to oral fluoropyrimidine drugs: A phase II trial on the individualization of postoperative adjuvant chemotherapy using oral fluorinated pyrimidines in stage III colorectal cancer treated by curative resection (ACT-01 Study). Oncology Reports 29, 437–444 (2013). 37. Lévi, F. & Okyar, A. Circadian clocks and drug delivery systems: impact and opportunities in chronotherapeutics. Expert opinion on drug delivery 8, 1535–41 (2011). 27 38. Lévi, F., Okyar, A., Dulong, S., Innominato, P. F. & Clairambault, J. Circadian timing in cancer treatments. Annual review of pharmacology and toxicology 50, 377–421 (2010). 39. Roenneberg, T. & Merrow, M. Entrainment of the human circadian clock. Cold Spring Harbor symposia on quantitative biology 72, 293–9 (2007). 40. Antoch, M. P. & Kondratov, R. V Pharmacological modulators of the circadian clock as potential therapeutic drugs: focus on genotoxic/anticancer therapy. Handbook of experimental pharmacology 217, 289–309 (2013). 41. Ortiz-Tudela, E., Mteyrek, A., Ballesta, A., Innominato, P. F. & Lévi, F. Cancer chronotherapeutics: experimental, theoretical, and clinical aspects. Handbook of experimental pharmacology 217, 261–88 (2013). 42. Granda, T. G. & Lévi, F. Tumor-based rhythms of anticancer efficacy in experimental models. Chronobiology international 19, 21–41 (2002). 43. Longley, D. B., Harkin, D. P. & Johnston, P. G. 5-fluorouracil: mechanisms of action and clinical strategies. Nature reviews. Cancer 3, 330–8 (2003). 44. Granda, T. G. et al. Circadian regulation of cell cycle and apoptosis proteins in mouse bone marrow and tumor. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 19, 304–6 (2005). 45. Pestalozzi, B. C. et al. Increased thymidylate synthase protein levels are principally associated with proliferation but not cell cycle phase in asynchronous human cancer cells. British journal of cancer 71, 1151–7 (1995). 46. Burns, E. R., Bagwell, C. B., Hinson, W. G., Pipkin, J. L. & Hudson, J. L. Preparation and stability of sixteen murine tissues and organs for flow cytometric cell cycle analysis. Cytometry 4, 150–60 (1983). 47. Wood, P. A. & Hrushesky, W. J. Circadian rhythms and cancer chemotherapy. Critical reviews in eukaryotic gene expression 6, 299–343 (1996). 48. Innominato, P. F., Lévi, F. A. & Bjarnason, G. A. Chronotherapy and the molecular clock: Clinical implications in oncology. Advanced drug delivery reviews 62, 979–1001 (2010). 49. Smaaland, R. et al. DNA synthesis in human bone marrow is circadian stage dependent. Blood 77, 2603–11 (1991). 50. Lévi, F., Zidani, R. & Misset, J. L. Randomised multicentre trial of chronotherapy with oxaliplatin, fluorouracil, and folinic acid in metastatic colorectal cancer. International Organization for Cancer Chronotherapy. Lancet 350, 681–6 (1997). 28 51. Abolmaali, K. et al. Circadian variation in intestinal dihydropyrimidine dehydrogenase (DPD) expression: a potential mechanism for benefits of 5FU chrono-chemotherapy. Surgery 146, 269– 73 (2009). 52. Krugluger, W. et al. Regulation of genes of the circadian clock in human colon cancer: reduced period-1 and dihydropyrimidine dehydrogenase transcription correlates in high-grade tumors. Cancer research 67, 7917–22 (2007). 53. Levi, F. & Schibler, U. Circadian rhythms: mechanisms and therapeutic implications. Annual review of pharmacology and toxicology 47, 593–628 (2007). 54. Bjarnason, G. A. & Jordan, R. Rhythms in human gastrointestinal mucosa and skin. Chronobiology international 19, 129–40 (2002). 55. Smaaland, R., Sothern, R. B., Laerum, O. D. & Abrahamsen, J. F. Rhythms in human bone marrow and blood cells. Chronobiology international 19, 101–27 (2002). 56. Kobayashi, Y. et al. Enzymatic activities of uridine and thymidine phosphorylase in normal and cancerous uterine cervical tissues. Human cell 20, 107–10 (2007). 57. Ruddy, K., Mayer, E. & Partridge, A. Patient adherence and persistence with oral anticancer treatment. CA: a cancer journal for clinicians 59, 56–66 58. Baraldo, M. The influence of circadian rhythms on the kinetics of drugs in humans. Expert opinion on drug metabolism & toxicology 4, 175–92 (2008). 59. Soran, A. et al. Centralized medical monitoring in phase III clinical trials: the National Surgical Adjuvant Breast and Bowel Project (NSABP) experience. Clinical trials (London, England) 3, 478– 85 (2006). 60. Raymond, E., Chaney, S. G., Taamma, A. & Cvitkovic, E. Oxaliplatin: a review of preclinical and clinical studies. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO 9, 1053–71 (1998). 61. Boughattas, N. A. et al. Circadian rhythm in toxicities and tissue uptake of 1,2diamminocyclohexane(trans-1)oxalatoplatinum(II) in mice. Cancer research 49, 3362–8 (1989). 62. Caussanel, J. P. et al. Phase I trial of 5-day continuous venous infusion of oxaliplatin at circadian rhythm-modulated rate compared with constant rate. Journal of the National Cancer Institute 82, 1046–50 (1990). 63. Lévi, F. et al. A chronopharmacologic phase II clinical trial with 5-fluorouracil, folinic acid, and oxaliplatin using an ambulatory multichannel programmable pump. High antitumor effectiveness against metastatic colorectal cancer. Cancer 69, 893–900 (1992). 64. Lévi, F. A. et al. Chronomodulated versus fixed-infusion-rate delivery of ambulatory chemotherapy with oxaliplatin, fluorouracil, and folinic acid (leucovorin) in patients with 29 colorectal cancer metastases: a randomized multi-institutional trial. Journal of the National Cancer Institute 86, 1608–17 (1994). 65. F. Levi, P. Innominato, A. Poncet, T. Moreau, S. Iacobelli, C. Focan, C. Garufi, G. Bjarnason, R. A. Meta-analysis of gender effect for first-line chronomodulated 5-fluorouracil-leucovorinoxaliplatin (ChronoFLO) compared with FOLFOX or constant infusion (conventional delivery, CONV) against metastatic colorectal cancer (MCC) in three international contr. 45th Annual Meeting of the American-Society-of-Clinical-Oncology, Orlando (2009). 66. Giacchetti, S. et al. Phase III trial comparing 4-day chronomodulated therapy versus 2-day conventional delivery of fluorouracil, leucovorin, and oxaliplatin as first-line chemotherapy of metastatic colorectal cancer: the European Organisation for Research and Treatment of Can. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 24, 3562–9 (2006). 67. Twelves, C. Capecitabine as first-line treatment in colorectal cancer. Pooled data from two large, phase III trials. European journal of cancer (Oxford, England: 1990) 38 Suppl 2, 15–20 (2002). 68. Cassidy, J. et al. Randomized phase III study of capecitabine plus oxaliplatin compared with fluorouracil/folinic acid plus oxaliplatin as first-line therapy for metastatic colorectal cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 26, 2006– 12 (2008). 69. Santini, D. et al. Continuous infusion of oxaliplatin plus chronomodulated capecitabine in 5fluorouracil- and irinotecan-resistant advanced colorectal cancer patients. Oncology 69, 27–34 (2005). 70. Reigner, B., Blesch, K. & Weidekamm, E. Clinical pharmacokinetics of capecitabine. Clinical pharmacokinetics 40, 85–104 (2001). 71. Santini, D. et al. Phase I study of intermittent and chronomodulated oral therapy with capecitabine in patients with advanced and/or metastatic cancer. BMC cancer 6, 42 (2006). 72. Santini, D. et al. Chronomodulated administration of oxaliplatin plus capecitabine (XELOX) as first line chemotherapy in advanced colorectal cancer patients: phase II study. Cancer chemotherapy and pharmacology 59, 613–20 (2007). 73. Qvortrup, C. et al. Chronomodulated capecitabine in combination with short-time oxaliplatin: a Nordic phase II study of second-line therapy in patients with metastatic colorectal cancer after failure to irinotecan and 5-flourouracil. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO 19, 1154–9 (2008). 74. Pfeiffer, P., Hahn, P. & Jensen, H. A. Short-time infusion of oxaliplatin (Eloxatin) in combination with capecitabine (Xeloda) in patients with advanced colorectal cancer. Acta oncologica (Stockholm, Sweden) 42, 832–6 (2003). 30 75. Pfeiffer, P. et al. Short-time infusion of oxaliplatin in combination with capecitabine (XELOX30) as second-line therapy in patients with advanced colorectal cancer after failure to irinotecan and 5fluorouracil. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO 17, 252–8 (2006). 76. Qvortrup, C. et al. A randomized study comparing short-time infusion of oxaliplatin in combination with capecitabine XELOX(30) and chronomodulated XELOX(30) as first-line therapy in patients with advanced colorectal cancer. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO 21, 87–91 (2010). 77. Tabata, T. et al. Bioactivation of capecitabine in human liver: involvement of the cytosolic enzyme on 5’-deoxy-5-fluorocytidine formation. Drug metabolism and disposition: the biological fate of chemicals 32, 762–7 (2004). 78. Lemmer, B. & Nold, G. Circadian changes in estimated hepatic blood flow in healthy subjects. British journal of clinical pharmacology 32, 627–9 (1991). 79. Chansky, K., Benedetti, J. & Macdonald, J. S. Differences in toxicity between men and women treated with 5-fluorouracil therapy for colorectal carcinoma. Cancer 103, 1165–71 (2005). 80. Lévi, F. et al. Implications of circadian clocks for the rhythmic delivery of cancer therapeutics. Advanced drug delivery reviews 59, 1015–35 (2007). 81. Bressolle, F. et al. Circadian rhythm of 5-fluorouracil population pharmacokinetics in patients with metastatic colorectal cancer. Cancer chemotherapy and pharmacology 44, 295–302 (1999). 82. Monteleone, P., Esposito, G., La Rocca, A. & Maj, M. Does bright light suppress nocturnal melatonin secretion more in women than men? Journal of neural transmission. General section 102, 75–80 (1995). 83. Burke, H. M., Davis, M. C., Otte, C. & Mohr, D. C. Depression and cortisol responses to psychological stress: a meta-analysis. Psychoneuroendocrinology 30, 846–56 (2005). 84. Louie, S. G. et al. Higher capecitabine AUC in elderly patients with advanced colorectal cancer (SWOGS0030). British journal of cancer 109, 1744–9 (2013). 85. Le Couteur, D. G. & McLean, A. J. The aging liver. Drug clearance and an oxygen diffusion barrier hypothesis. Clinical pharmacokinetics 34, 359–73 (1998). 86. Etienne-Grimaldi, M.-C. et al. Chronopharmacokinetics of oral tegafur and uracil in colorectal cancer patients. Clinical pharmacology and therapeutics 83, 413–5 (2008). 87. Bruguerolle, B. Chronopharmacokinetics. Current status. Clinical pharmacokinetics 35, 83–94 (1998). 88. Reinberg, A. & Smolensky, M. H. Circadian changes of drug disposition in man. Clinical pharmacokinetics 7, 401–20 31 89. Moore, J. G. & Englert, E. Circadian rhythm of gastric acid secretion in man. Nature 226, 1261–2 (1970). 90. Tabata, T. BIOACTIVATION OF CAPECITABINE IN HUMAN LIVER: INVOLVEMENT OF THE CYTOSOLIC ENZYME ON 5’-DEOXY-5-FLUOROCYTIDINE FORMATION. Drug Metabolism and Disposition 32, 762–767 (2004). 91. Czejka, M., Schueller, J., Farkouh, A., Gruenberger, B. & Scheithauer, W. Plasma disposition of capecitabine and its metabolites 5’DFCR and 5'DFUR in a standard and dose-intensified monotherapy regimen. Cancer chemotherapy and pharmacology 67, 613–9 (2011). 92. Mercier, C. et al. Early severe toxicities after capecitabine intake: possible implication of a cytidine deaminase extensive metabolizer profile. Cancer chemotherapy and pharmacology 63, 1177–80 (2009). 93. Mori, K. et al. Expression levels of thymidine phosphorylase and dihydropyrimidine dehydrogenase in various human tumor tissues. International journal of oncology 17, 33–8 (2000). 94. Gamelin, E. et al. Individual fluorouracil dose adjustment based on pharmacokinetic follow-up compared with conventional dosage: results of a multicenter randomized trial of patients with metastatic colorectal cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 26, 2099–105 (2008). 95. Deenen, M. J., Rosing, H., Hillebrand, M. J., Schellens, J. H. M. & Beijnen, J. H. Quantitative determination of capecitabine and its six metabolites in human plasma using liquid chromatography coupled to electrospray tandem mass spectrometry. Journal of chromatography. B, Analytical technologies in the biomedical and life sciences 913-914, 30–40 (2013). 32