RNA-seq_method_1
advertisement

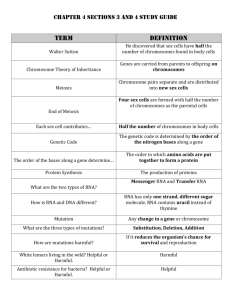
RNA-seq method 1 Kits used: Microbexpress – life tech NEBNext Magnesium RNA Fragmentation module – NEB NEBNext small RNA library prep module – NEB Overview: Total RNA isolation from cells Removal of rRNA from total Fragmentation of small RNA (including mRNA, microRNA, etc.) Purification Kinase treatment Purification QC - Agilent trace to verify correct sizing 3’ ligation Primer hybridization 5’ ligation First strand synthesis PCR Amp Size selection QC – Agilent trace Send for Illumina HiSeq sequencing Total RNA isolation from cells RNAsnap method RNA extraction solution (needs to be made fresh before each use). • 18mM EDTA • 0.025% SDS • 1% 2-mercaptoethanol • 95% formamide (RNA grade) Protocol: For bacteria culture composed of 10^8 cells. Centrifuge 1 ml of culture at 16 000g for 30s and remove supernatant. Store cell pellet in dry ice until ready for extraction. Resuspend cell pellet in 100ul of RNA extraction solution by vigorously vortexing. Incubate sample at 95 C for 7min to lyse cells. Pellet the warm sample by centrifuging at 16 000g for 5min at room temperature. Transfer supernatant containing RNA to fresh tube without disturbing the clear gelatinous pellet. Quantify the RNA based on A260. First use the RNA extraction solution as the blank. According to paper, a one ml sample of an early exponential culture of E. coli (10^8 cells) yielded 60 +- 3 ug of total RNA. Perform RNA clean-up and DNase I treatment using Zymo RNA Clean and Concentrator -25 kit. RNA clean-up (>17nt total RNA) with In-column DNase Digestion Add 2 volumes of RNA Binding Buffer to each volume of RNA sample (minimum vol is 50ul) and mix well. Add 1 volume ETOH (95-100%) to the mixture and mix well. Transfer the mixture to the Zymo-Spin IIC column in a collection tube and centrifuge at >12,000 x g for 1 minute. Discard the flow through. Make the following DNase I cocktail (for each sample add) RNase-Free DNase I 10ul (1U/ul) 10x Reaction Buffer 10ul RNA Wash buffer 80ul Add 400ul of RNA wash buffer to the Zymo-Spin IIC column in a collection tube and centrifuge at >12,000 x g for 30 seconds. Discard the flow through. Add 100ul DNase I cocktail directly to the matrix of the Zymo-Spin IIC column. Incubate the column at 25-370C for > 15 minutes (optimal temp for DNase I is 370C). Centrifuge > 12,000 x g for 30 seconds. Discard the flow through. Add 400ul RNA Prep Buffer to the column and centrifuge at 12,000 x g for 1 minute. Discard the flow trough. Add 800ul RNA Wash Buffer to the column and centrifuge at > 12,000 x g for 30 seconds. Discard the flow through. Repeat this wash step with 400ul RNA Wash Buffer. Centrifuge the Zymo-Spin IIC Column in an emptied collection tube at >12,000 x g for 2 minutes. Remove the column carefully from the collection tube and transfer to new RNase-free tube. Add >25 ul of DNase/RNase-Free water directly to the column matrix and let stand for 1 minute at room temperature. Centrifuge at 10,000 x g for 30 seconds. Use the eluted RNA immediately or store at -80. Removal of rRNA from total RNA MICROBExpress kit General notes: A260:A280 should be > 1.7. The volume of RNA should be less than 15ul. Recommended max amount of RNA per rxn is 10ug. Can work with as little as 1ug or less RNA. Procedure: Part A (Anneal RNA and Capture Oligonucleotide Mix) Pipet 200ul of Binding Buffer into a 1.5ml tube. Add total RNA (2-10ug in max vol of 15ul) to the Binding Buffer. Close tube and tap or gently vortex to mix. Add 4ul of Capture Oligo Mix to the RNA. Close tube and tap or gently vortex to mix, microfuge briefly to get mixture to bottom of tube. Incubate the mixture at 700C for 10 minutes. (This will denature secondary structures in RNA including 16S and 23S ribosomes and helps to facilitate maximal hybridization of the rRNAs to the capture oligonucleotides). Anneal at 370C for 15 minutes. Part B (Prepare the Oligo MagBeads)…must vortex the magbead tube before pipetting. For each RNA sample, transfer 50ul of Oligo MagBeads to a 1.5ml tube. Beads for up to 10 RNA samples can be processed in a single 1.5ml tube. Place the tube in a magnetic stand, leave on stand for ~ 3 minutes. Carefully remove and discard the supernatant. Add Nuclease-free water to the beads; use a volume of water equal to the original volume of beads used. Remove the tube from the magnetic stand and resuspend the beads by brief, gentle vortexing. Place the tube in a magnetic stand, leave on stand for ~ 3 minutes. Carefully remove and discard the supernatant leaving the beads in the tube. Add binding buffer to the beads; use volume equal to the original volume of MagBeads used. Remove the tube from the magnetic stand and resuspend the beads by brief, gentle vortexing. Place the tube in a magnetic stand, leave on stand for ~ 3 minutes. Carefully remove and discard the supernatant leaving the beads in the tube. Add fresh binding buffer to the beads; use volume equal to the original volume of MagBeads used. Remove the tube from the magnetic stand and resuspend the beads by brief, gentle vortexing. Place the Oligo MagBead slurry in a 370C incubator and allow the temperature to equilibrate to 370C before proceeding. Part C (Capture the rRNA and recover the enriched mRNA) Heat the Wash Solution to 370C. Gently vortex the Oligo MagBead tube to resuspend the beads. Add 50ul of the beads to the RNA/Capture Oligo Mix from Part A. Very gently vortex or tap the tube to mix and microfuge very briefly to get a mixture to the bottom of tube. Incubate 15 minutes at 370C. Place the tube on magnetic stand., leave on stand for ~ 3 minutes. Remove the supernatant which contains the enriched mRNA, being careful not to dislodge the beads. Transfer to a collection tube on ice. Add 100ul of prewarmed Wash Solution to the beads. Remove the tube from the magnetic stand and resuspend the beads by brief, gentle vortexing. Place the tube on magnetic stand., leave on stand for ~ 3 minutes. Remove the supernatant which contains the enriched mRNA, being careful not to dislodge the beads. Pool which the RNA already collected. Part D (Precipitate and Resuspend the Enriched mRNA) Protocol recommends ethanol precipitation. Going to try Zymo RNA clean and concentrator in tandem and compare the two methods. Assess yield using Aglient trace and/or Nano drop. Fragmentation of RNA NEBNext Magnesium RNA Fragmentation Module Protocol (E6150) Protocol: Starting material: Total RNA (2-50ug) or purified mRNA (50-250ng) Mix the following components in a sterile PCR tube: RNA 1-18 ul RNA frag buffer 10x 2ul Nuclease-free water x Total volume is 20 ul Incubate in a preheated thermal cycler for 4 mins at 940C. Transfer tube to ice. Add 2 ul 10x RNA Fragmentation stop solution. Clean-up using Zymo RNA clean –concentrate kit. Kinase treat fragmented RNA Mix the following components in a sterile PCR tube Fragmented RNA 18ul T4 PNK Buffer, 10x 2.0ul T4 PNK 2.0ul ATP, 10mM 2.0ul Total volume 24ul Flick tubes to mix, briefly spin tubes down. Incubate at 370C for 30 minutes. Snap-cool on ice. Clean-up using Zymo RNA clean –concentrate kit. QC step: send 2 ul to GSAF to be run on RNA pico or nano chip. Median size should be 100-200nts. If trace looks go proceed to library prep. Library Preparation method 1 NEBNext Small RNA Library Prep Set for Illumina (Multiplex Compatible) E7330S/L (24/96 rxns) Starting Material: 100ng – 1ug RNA Step 1: Ligate the 3’ SR Adaptor Note: for RNA inputs of 100ng, dilute the 3’ SR Adaptor for Illumina 1:2 Mix the following components in a sterile nuclease-free PCR tube: Input RNA 1-6ul 3’ SR adaptor 1 ul Nuclease-free water x Total volume 7ul Incubate in a preheated thermal cycler for 2 minutes at 700C. Transfer tube to ice. Add the following to tube: 3’ ligation reaction buffer (2x) 10ul 3’ ligation enzyme mix 3ul Total volume (7+13) 20ul Incubate for 1 hour at 250C. Step 2: Hybridize the RT (reverse transcription primer) Add the following to ligated material from step 1: Nuclease-free water 4.5ul SR RT Primer 1 ul Total volume (20 +5.5) 25.5ul Heat sample (3 different times and temperatures): 1. 5 min at 750C 2. 15 min at 370C 3. 15 min at 250C Step 3: Ligate the 5’ SR Adaptor First the 5’ adaptor must be denatured With 5 minutes remaining from last heat step, resuspend the 5’ SR Adaptor in 120ul of nuclease free water. Aliquot 1.1n ul of the 5’ adaptor into a separate, 200ul PCR tube, with n = number of samples being processed. Incubate the 5’ adaptor in thermal cycler at 700C for 2 minutes. Immediately place the tube on ice and use within 30 minutes. Add the following to ligated material from step 2: 5’ SR Adaptor for Illumina (denatured) 5’ ligation reaction buffer (10x) 5’ ligation enzyme mix Total volume (25.5 + 4.5) 1ul 1ul 2.5ul 30ul Incubate for 1 hour at 250C. Step 4: Preform RT reaction Mix the following components in a sterile nuclease-free PCR tube: Adaptor ligated RNA from step 3 30ul First strand synthesis buffer 8 ul Murine RNase Inhibitor 1 ul M-MuLV Reverse Transcriptase (RNase H-) 1 ul Total volume 40ul Incubate for 1 hour at 500C. This is a safe stopping point; if stopping at this point incubate the RT reaction at 700C for 15 minutes to inactivate the transcriptase. Step 5: PCR amplification Add the following to the RT reaction mix from step 4: LongAmp Taq 2X master mix SR primer for Illumina Index (x) primer * Nuclease free water Total volume (40 +60) 50ul 2.5ul 2.5ul 5.0ul 100ul * Use different index primer for each sample (contains bar code). Use either our in-house primers or NEB Illumina adaptors. PCR Cycling conditions Cycle Step Initial denaturation Denaturation Annealing Extension Final Extension Hold TEMP 940 C Time 30 sec Cycles 1 940 C 620 C 700 C 700 C 40 C 13 sec 30 sec 15 sec 5 min - 12-15 12-15 12-15 1 Step 6: Size selection using AMPure XP beads Transfer 100ul of sample to a 1.5ml tube. Add 120ul (1.3X) or resuspended AMPure XP beads and mix well. Incubate for 5 minutes at room temperature. Place the tube on magnetic stand and allow to stand for 5 minutes. After solution is clear, carefully transfer the supernatant (230ul) to a new tube (DO NOT DISCARD). Discard the tubes containing the beads and thus the large DNA fragments. Add 370ul (3.7X) of resuspended AMPure XP beads and mix well. Incubate for 5 minutes at room temperature. Place the tube on magnetic stand for 5 minutes. After solution is clear, carefully remove and discard the supernatant. Add 1ml of freshly prepared 80% ethanol to the tube on the mag stand and incubate for 30 secs. Repeat wash step once. Briefly spin tube and return the magnetic stand. Completely remove the residual ethanol and air-dry the beads for 10 minutes. Elute the DNA target from the beads with 15 ul of nuclease-free water. Run 1ul on Bionalyzer High Sensitivity chip to check for correct size. Step 7: Submit sample to GSAF for Illumina Hi-seq sequencing • • GSAF requests 20ul of > 1nM for Illumina HiSeq. Minimum recommended amount is 0.001 picomoles per 1 million reads, >50pM.