supplemental materials and methods (text)
advertisement
")
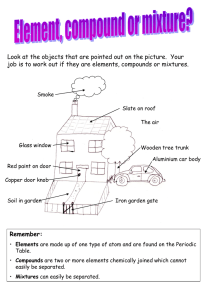
1 SUPPLEMENTAL MATERIALS AND METHODS (TEXT) INTERSPECIFIC COMPARISON OF CONSTITUTIVE ASH PHLOEM PHENOLIC CHEMISTRY REVEALS COMPOUNDS UNIQUE TO MANCHURIAN ASH, A SPECIES RESISTANT TO EMERALD ASH BORER JUSTIN G.A. WHITEHILL1,2*, STEPHEN O. OPIYO3, JENNIFER L. KOCH4, DANIEL A. HERMS5, DONALD F. CIPOLLINI6, AND PIERLUIGI BONELLO1 1 2 3 Michael Smith Laboratories, University of British Columbia, 301-2185 East Mall, Vancouver, BC, Canada V6T 1Z4 Molecular and Cellular Imaging Center-Columbus, Ohio Agricultural Research and Development Center, 2021 Coffey Road, Columbus, OH 43210 4 5 Department of Plant Pathology, The Ohio State University, 2021 Coffey Road, Columbus, OH 43210 Northern Research Station, USDA Forest Service, 359 Main Road, Delaware, OH 43015 Department of Entomology, The Ohio State University, Ohio Agricultural Research and Development Center, 1680 Madison Ave., Wooster, Ohio 44691 6 Department of Biological Sciences, Wright State University, 3640 Colonel Glenn Highway, Dayton, OH 45435 Justin G. A. Whitehill () Michael Smith Laboratories The University of British Columbia 301-2185 East Mall Vancouver, BC V6T 1Z4 Canada E-mail: whiteh5@msl.ubc.ca 2 Selection of Phenolic Compounds Using HPLC-UV for Qualitative, Quantitative, and Statistical Analyses - HPLC-UV analyses of phenolic extracts were performed on an Alliance 2690 separation module (Waters, Milford, MA, USA) equipped with an autosampler and a 996 Photodiode Array Detector (Waters). The autosampler and column temperatures were set to 4 and 30º C, respectively. Chromatographic separations were accomplished using a Waters Xterra RP18, 5 μm, 4.6×150 mm column and a Waters Xterra RP18, 3.9 μm, 3.0 × 20 mm guard column. The binary mobile phase consisted of water/acetic acid (A) (98:2, v/v) and methanol/acetic acid (B) (98:2, v/v), with a flow rate of 1 ml/min. The elution program followed that of Eyles et al. 2007. The injection volume for all samples was 10 μl. Samples were passed through a Photodiode Array Detector (PDA) (scanning range, 200-400 nm) with individual peaks quantified at 280 nm. The absorbance limit for detection was set to a minimum peak area of 9,000 in the processing parameters prior to data export. Individual peaks were included in subsequent statistical analyses if they met the following criteria: 1) a peak height greater than 0.02 AU, 2) a well defined UV spectrum, and 3) were consistently detected in at least three individual trees within a taxon. Analysis of Soluble Phenolics with HPLC-ESI-MS-PDA - For qualitative identification of phenolic compounds in phloem extracts, a pooled sample was generated for each taxon on both sampling dates by combining equal amounts of phenolic extract (100 μl) from each replicate tree sampled, for a total of 14 sample pools (7 taxa x 2 sampling dates). Pooled samples were used only in qualitative analyses, while samples from each individual replicate tree were used for all quantitative analysis. For each sampling date, 3 the pooled extract from each taxon was analyzed using an HPLC-ESI-MS (Varian 500 MS; Palo Alto, CA, USA) in parallel with a PDA detector (Varian ProStar 335). Chromatographic separations were accomplished using a Waters Xterra RP18, 5 μm, 4.6×150 mm column. The binary mobile phase consisted of water/acetic acid (99.9:0.1 v/v) (eluant A) and methanol/acetic acid (99.9:0.1 v/v) (eluant B) with a flow rate of 1 ml/min. The gradient was as follows (percentages refer to proportions of eluant B): 515% (0-15 min); 15-30% (15-35 min); 30-40% (35-40 min); 40-60% (40-50 min); 6090% (50-55 min); 90-100% (55-60 min). The injection volume for all samples was 10 μl. Separation of compounds was achieved by post-column splitting (1:1) where half of the LC effluent was passed through the PDA Detector (scanning range, 200–400 nm), and the other half through an electrospray source (parameters below). The ESI was operated in both the negative and positive ion modes. The MS detector was optimized to obtain maximum yields of [M-H]- ions of esculin, luteolin, rutin, oleuropein, tyrosol, and verbascoside standards. The optimized MS parameters were: capillary voltage, -80 V; needle voltage, -5 kV (for the negative ion mode; the parameters remained the same in the positive mode except the polarity was reversed). The atmospheric pressure ionization (API) parameters were as follows: API nebulizing gas (air) 25 psi; API drying gas (nitrogen) 15 psi at 350° C. Survey scan was set to detect molecules between 50 – 1000 m/z. All samples were analyzed using both the TurboDDS and full scan mode in negative and positive ion modes. The TurboDDS trigger threshold was set to 5,000 counts (parent ion counts). In the TurboDDS scan mode, MS scan parameters were set on-the-fly by the instrument to detect the most abundant parent ions and obtain maximum 4 yields of those compound fragments. If TurboDDS were triggered, the mode temporarily switched to MSn mode to perform daughter scans of the putative parent ions. The TurboDDS trigger threshold for daughter ions was 5,000, 500, and 50 ions for MS2, MS3, and MS4 respectively. Full scan parameters were set following the conditions established above. Full scan chromatograms were overlaid with PDA chromatogram traces at 280 nm to match [M-H]- parent ions to max of individual phenolic compounds. When compounds were not detected in the negative ion mode, the positive ion mode was used for identification (Table 1 and S1). PDA data of individual compounds run on the HPLC-ESI-MS-PDA were analyzed using PolyView software (Varian), and then matched to the max data generated from the UV analysis conducted on the Waters HPLC system in order to definitively identify peaks. Phenolic compounds were identified based on the congruence of parent and daughter ions, max, retention time, and order in which compounds eluted from the stationary phase (Table 1 and S1). Data acquisition and processing were performed using MS Workstation 6.9.2 (Varian). Identification and Quantification of Phenolics - The compounds identified from the phloem samples are reported in Table 1 and S1. Esculin (peak 12), esculetin (13), syringin (15), fraxin (18), fraxetin (22), 3-caffeoyl quinic acid (chlorogenic acid) (23), pinoresinol (39), verbascoside (45), oleuropein (51), quercetin diglycoside (rutin) and quercetin glycoside (isoquercetin) (55), luteolin (65), and apigenin (66) were unequivocally confirmed by spiking samples with the respective standards and matching retention times, UV maxima, and LC-MS characteristics. Esculin, esculetin, fraxin, fraxetin, 3-caffeoyl quinic acid (chlorogenic acid), verbascoside, oleuropein, quercetin 5 diglycoside (rutin), quercetin glycoside (isoquercetin), luteolin, and apigenin were purchased from Extrasynthese (Genay, France). Syringin was purchased from Apin Chemicals Limited (Oxfordshire, UK), and pinoresinol was purchased from Arbo Nova (Turku, Finland). External standards were lacking for other compounds, which were characterized and quantified following the methods of Eyles et al. (2007). Their concentrations were expressed in mg g-1 FW as: hydroxytyrosol equivalents for hydroxytyrosol hexoside (1); tyrosol equivalents for tyrosol hexoside (3) and tyrosol hexoside pentoside (11); vanillic acid equivalents for vanillic acid hexoside acetate adduct A (2), vanillic acid hexoside acetate adduct B (5), and unknown phenolic acid 1 (4); 3-caffeoyl-quinic acid (chlorogenic acid) equivalents for caffeoyl-quinic acid A (8); fraxetin equivalents for fraxetin related compound (6); esculetin equivalents for esculetin A (7), esculetin B (10), esculetin C (13), and unknown coumarin 1 (14); esculin equivalents for escuside (47); fraxidin equivalents for fraxidin A (16), fraxidin B (19), and mandshurin (21); fraxin equivalents for fraxin related compound (54), unknown coumarin 2 (59), and unknown coumarin 3 (60); syringin equivalents for unknown monolignol 1 (17); pinoresinol equivalents for unknown lignan 1 (24), pinoresinol diglucoside + 2H2O (25), (+)-1hydroxypinoresinol-4’-O-glucoside + 2H2O (28), unknown lignan 3 (30), and pinoresinol A (37); oleuropein equivalents for ligustroside A (27), oleuropein related compound 1 (29), unknown secoiridoid 1 (31), ligustroside B (33), 10-hydroxyoleuropein (36), dimethyloleuropein (38), oleuropein A (44), oleuropein B (48), demethylligstroside (49), oleuropein related compound 2 (56), ligustroside (57), and unknown secoiridoid 2 (62); verbascoside equivalents for lugrandoside A (26), calceolarioside C (32), β- 6 methoxylferrunginoside (40), calceolarioside A (42), lugrandoside B (43), verbascoside A (46), calceolarioside B (50), verbascoside B (53), unknown phenylethanoid 1 (63), and verbascoside C (64); apigenin for apigenin glucoside (58); and luteolin for kaempferol galactoside (61). We were unable to quantify elenolic acid derivative 1 (9), caffeoylshikimic acid (35), and elenolic acid derivative 2 (52) due to a lack of relevant external standards. We utilized both the negative and positive ion modes to maximize detection of coumarins in all of our pooled extracts. As previous studies have noted, the positive ion mode offers better sensitivity than the negative ion mode in detecting and identifying coumarins from crude plant extracts, and some coumarins were identified only in the positive ion mode (Yang et al. 2010). We identified three instances in which two compounds were co-eluting: (9) and (11), (19) and (20), (39) and (41) (Table 1 and S1; Figure 3). Peak (55) was a mix of rutin and quercetin, with neither dominating the peak. When fragments from the co-eluting compounds were detected in the same peak for June and August samples using LC-MS full scan mode, the more abundant compound is listed for that time in Table 1 and S1. TurboDDS mode was used to automatically detect compounds that were more abundant using on-the-fly parameters as specified above. Statistical Analyses - Principal component analysis (PCA) was used to investigate the overall relationships between phenolic metabolites and species through dimensionality reduction and feature extraction (Johnson and Wichern 1998). The PCA was conducted using R software (R Development Core Team, 2010) for both species and metabolites. PCA were run twice (Figure 4 and S1) in order to better visualize relationships between 7 species. The second PCA was based on data obtained from a cluster analysis of species and metabolites following the first PCA. Cluster analysis was run to support the grouping of variables obtained in the PCA. The cluster analyses were carried out using the “pvclust” routine in the pvclust package in R (Suzuki and Shimodaira 2006). The pvclust package generates approximately unbiased (AU) confidence values for the clusters using the bootstrap resampling technique to assess their reliability (Efron et al. 1996; Suzuki and Shimodaira 2006). A total of 1000 bootstrap replications were generated for each cluster, and the AU confidence values were used to assess the uncertainty. The significance level of the clusters was set to 95. A larger confidence value is indicative of a “true” cluster (Efron et al. 1996). In this study we also used biplots to identify the relationship between species and metabolites. Biplots were also constructed using R. Individual peak areas and mg g-1 FW concentrations for lignin were analyzed using univariate analysis of variance (ANOVA). Exploratory analyses of data and Levene’s test were used to evaluate variance equality and normality requirements of residuals. Square-root and logarithmic transformations were used to meet normality requirements of residuals and homogeneity of variance. Following significant F-tests, means were separated using the LSD test ( = 0.05). All data were analyzed with IBM SPSS Statistics v. 19 (SPSS Inc. 2010).