tpj12623-sup-0009-Legends
advertisement

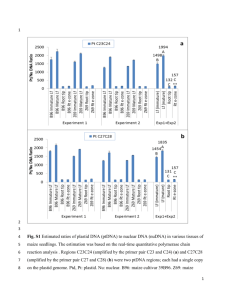
Supporting Information Legends Figure S1. Phylogenetic analysis of HAG co-regulators from maize. a), b), c), The ZmHAG family of GNC5-related histone acetyltransferases can be subdivided into three subfamilies named GCN5, ELP and HAT1 respectively (Liu et al., 2012). Each of the cloned HAG maize co-regulators was scanned for the presence of an acetyl-CoA binding site using the Conserved Domain database (CDD). This region (approximately 75 to 90 amino acids) was then used as the operational taxonomic unit for phylogenetic analysis. Related proteins were aligned using CLUSTALW using default parameters. The phylogenetic tree was exported in Newick format and displayed using FigTree V1.4.0. Raw branch lengths are shown to scale. Figure S2. Phylogenetic analysis of AUX/IAA co-regulators from maize. Full length amino acid sequences were aligned using MUSCLE using default parameters (VTML 200 substitution matrix with a gap opening penalty of -2.9 a gap extension penalty of 0). The maximum number of iterations was 8 with UPMGB as the cluster method and CLUSTALW for sequence weighting. The initial distance measure was Kmer 6-6 and Kimura % identity used for later iterations. The phylogenetic tree was exported in Newick format and displayed using FigTree V1.4.0. Raw branch lengths are shown to scale. Figure S3. Nucleotide sequence alignments showing the codon usage of the three synthetic codon optimized constructs for GRMZM2G162434, GRMZM2G001875, and GRMZM2G051793. Nucleotide sequences for the codon optimized constructs were retrieved from the sequence files provided by GeneArt®. Native sequences were retrieved from a CDS library of B73 RefGen_v2. Multiple alignments were then generated amongst the native, maize optimization, yeast optimization, and hybrid optimization CDS using SeqMan™ of the DNASTAR bioinformatics suite. 1 Figure S4. Workflow describing the cloning process used to generate the maize TFome collection. Table S1. RNA-Seq data used for the validation of gene models. Reads from 24 publically available RNA-Seq runs (Table S1a) and reads from 23 published and unpublished RNA-Seq runs (Table S1b) generated in our lab or through collaborations were used for our alignments. Reads from each sample were aligned to the B73 RefGen_v2 using Tophat. Next, each alignment file was merged using SAMtools. BEDtools was then used to intersect the merged alignment against the genomic coordinates of our genes of interest. The .bam files were then loaded into Integrative Genomics Viewer for visualization. Table S2. Table shows the percentage of clones successfully amplified from a variety of maize tissues by RT-PCR (n= 244). Table S3. Sequence discrepancies between maize TFome clones and B73 RefGen_v2 models. 2