Intermolecular and Surface Forces
advertisement

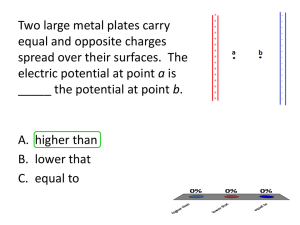
2.1 Intermolecular and Surface Forces ChemE 554/ove Intermolecular and Surface Forces 2.1.1 Overview: Types of Surface Forces There are three aspects that are of particular importance for any interaction: (a) its strength, (b) the distance over which it acts, and (c) the environment through which it acts. Strengths and distances for the most common intermolecular "bonds" are as follows: Nature of Bond Type of Force Ionic bond Coulombic force Covalent bond Electrostatic force (wave function overlap) Metallic bond Hydrogen Bond Van der Waals free valency electron sea interaction (sometimes also partially covalent (e.g., Fe and W) a strong type of directional dipole-dipole interaction (i) dipole-dipole force (ii) dipole-induced dipole force (iii) dispersion forces (charge fluctuation) Energy (kcal/mol) 180 240 170 283 26 96 210 (NaCl) (LiF) (Diamond) (SiC) (Na) (Fe) (W) 7 (HF) 2.4 (CH4) Distance 2.8 Å 2.0 Å N/A 4.3 Å 2.9 Å 3.1 Å significant in the range of a few Å to hundreds of Å The integral form of interaction forces between surfaces of macroscopic bodies through a third medium (e.g., vacuum and vapor) are named surfaces forces. Table 1 provides an overview of the types of surface forces. One differentiates between short range (e.g., Van der Waals interaction) and long range surface forces (e.g., electromagnetic interactions). Combinations of interactions lead to new forces such as the DLVO forces. In vacuum, the main contributors to long-range surface interactions are the Van der Waals and electromagnetic interactions. At separation distance < 2 nm one might have also to consider short range retardation due to covalent or metallic bonding forces. Van der Waals and electromagnetic interactions can be both, attractive or repulsive. In the case of a vapor environment as the third medium (e.g., atmospheric air containing water and organic molecules), one has also to consider modifications by the vapor due to surface adsorption or interaction shielding. This can lead to force modification or additional forces such as the strong attractive capillary forces. 1 2.1 Intermolecular and Surface Forces ChemE 554/ove Source: Handbook of Micro/Nanotribology, ed. Bharat Bhushan, CRC Press N.Y. p. 269 (1995). 2 2.1 Intermolecular and Surface Forces ChemE 554/ove Covalent Bond: The standard example for a covalent bond is the hydrogen atom. When the wave-function overlap is considerable, the electrons of the hydrogen atoms will be indistinguishable. The total energy will be decreased by the "exchange energy", which causes the attractive force. The characteristic property of covalent bonds is a concentration of the electron charge density between the two nuclei. The force is strongly directed and falls off within a few Ǻngstroms. Ionic Bonds: These are simple Coulombic forces which are a result of the electron transfer. For example in lithium fluoride the lithium transfers its 2s-electron to the fluorine 2p-state. Consequently the shells of the atoms are filled up, but the lithium has a net positive charge and the fluorine a net negative charge. These ions attract each other by Coulombic interaction which stabilizes the ionic crystal in the rock-salt structure. Metallic Bonds and Interaction: The strong metallic bonds are only observed when the atoms are condensed in a crystal. They originates from the free valency electron sea which holds together the ionic cores. A similar effect is observed when two metallic surfaces approach each other. The electron clouds have the tendency to spread out, in order to minimize the surface energy. Thus a strong exponentially decreasing, attractive interaction is observed. 2.1.2 Capillary Forces Capillary forces are meniscus forces due to condensation. It is well known that micro-contacts act as nuclei of condensation. In air, water vapor plays the dominant role. If the radius of curvature of the micro-contact is below a certain critical radius a meniscus will be formed. This critical radius is defined approximately by the size of the Kelvin radius rK = l/(l/rl + 1/r2) where rl and r2 are the radii of curvature of the meniscus. The Kelvin radius is connected with the partial pressure ps (saturation vapor pressure) by LV rK p RT log ps where L is the surface tension, R the gas constant, T the temperature, V the mol volume and p/ps the relative vapor pressure (relative humidity for water). The surface tension L of water is 0.074N/m (T=20°C) leading to a critical Van der Waals distance of water of LV/RT = 5.4 Å. Consequentially, we obtain for p/ps=0.9 a Kelvin radius of 100 Å. At small vapor pressures, the Kelvin radius gets comparable to the dimensions of the molecules, and thus, the Kelvin equation breaks down. The meniscus forces between two objects of spherical and planar geometry can be approximated, for D « R, as: 4R L cos F R D 1 D / d where R is the radius of the sphere, d the length of PQ , see Fig. 1, D the distance between the sphere and the plate, and the meniscus contact angle. 3 2.1 Intermolecular and Surface Forces ChemE 554/ove Fig. 1: Capillary meniscus between two two objects of spherical and planar geometry The maximum force, found at at D = 0 (contact), is R d Fmax 4R cos . Capillary Neck in Nanocontacts Pull-off Force The medium for capillary interaction is the capillary neck. Structured bulk water strongly affects the surface tension of the water-air interface, i.e., the mechanical properties of neck side-walls. At the water-solid interface, the water experiences surface adhesion that competes with the molecular self-association of bulk water. At sufficiently low humidity, i.e., in a spatially confined liquid film of only a few molecular layers, it can be expected that the interfacial interaction is powerful enough to distort the bulk structure. Salmeron and co-workers employed SFM adhesion measurements on mica surfaces as a function of the humidity and noticed that there are three distinct force regimes as illustrated in Fig. 2 (experimental confirmation provided in Fig. 3). In Regime I, the measured pull-off forces are depressed if compared to the forces in Regime II and III. The qualitative force behavior from regime I to II has been confirmed by others with hydrophilic SFM tips on mica.(10,14) In order to reflect on the possibility that the qualitative transition behavior resembles structural change of water, it has first to be discussed on how a structural change would affect the observable force. I II III Fig. 2: Generic sketch of the functional relationship between the pull-off force and the relative humidity (RH). Regimes I, II and III represent the van der Waals regime, mixed van der Waals – capillary regime, and capillary regime decreased by repulsive forces, respectively. Relative Humidity 4 2.1 Intermolecular and Surface Forces ChemE 554/ove Figure 3 shows the results of pull-off force vs. RH measurements conducted with a hydrophilic tip on a silicon sample. At low RH (40%), the pull-off force is constant. In the mid-RH range (40%RH70%), the pull-off force increases with increasing RH. A pull-off force RH hysteresis is noticeable in this regime. At 40% RH a force discontinuity occurs. The transition seems to be more pronounced for decreasing humidity than for increasing humidity, which is an instrumental artifact due to improved control of RH for decreasing humidity. At RH larger than 70%, the pull-off force decreases with increasing humidity. The transition RH does not change with spring constants. Fig. 3: Pull-off force vs. RH measured between a hydrophilic tip and a flat silicon sample. measured when increasing RH, measured when decreasing RH. Pull-off Force (nN) 40 30 20 10 0 0 20 40 60 80 100 Relative Humidity (%) A qualitative and quantitative similar results was found for a "macroscopic" silica glass sphere cantilever (microcontact). There, the pull-off force stepped up at around 3040% RH.. Typically, the capillary force of bulk water is estimated by the following equation, assuming a sphere-plane geometry (Fig. 1), R d (1) Fcap 4R cos (see derivative below for nanocontacts), where R is the radius of the sphere, d the length of PQ , the liquid surface tension, and the meniscus contact angle.(25) Note that the capillary force described by Eq. (1) is only dependent on the surface tension of bulk water and the contact angle , but is independent of the solid-liquid and solid-solid interaction parameters. Equation (1) predicts a gradual change in the capillary force with the meniscus contact angle. This equation does not explain the force transition experimentally observed (as depicted in Fig. 2) The dilemma seems to be solved if one assumes that the force instability at around 40 % RH reflects a structural transition of water, i.e., is not a constant, but changes at ~40% RH. Note that the thickness of condensed water vapor film is strongly related to RH, thus a boundary regime at the solid surface could be defined in which water undergoes a structural change. Tthis hypothesis is plausible for a highly ordered mica stubstrate, but raises suspicion in the case of unstructured silicon-oxide surface. Let us assume that water undergoes a phase change at 40% RH for hydrophilic silicon samples. This phase change can be assumed to be independent of pressure confinement, otherwise the transition for a sharp tip and a microsphere, (Figs. 3), would have occurred 5 2.1 Intermolecular and Surface Forces ChemE 554/ove at significantly different RH values. The thickness of the water film on the substrate surface depends on RH. Thus, the restructuring transition in water occurs in the vicinity closest to the silicon substrate, because the water film is thinning with decreasing humidity. Note that only one hydrophilic surface is necessary to form a water film. Hence, the water restructuring process and its detection in pull-off force measurements should not depend on the cantilever probe material as long as the sample is hydrophilic. For a hydrophobic tip coated with n-octadecyltrichlorosilane (OTS), on the same silicon substrate as above, one observed however constant pull-off forces (i.e., forces independent of RH) in the entire range from 10% to 80% RH (Fig. 4). Consequently the water structuring model based on force-distance curves is inconsistent. A much more likely interpretation for the force instability at 40% RH is the ability or inability of the water film to form a liquid joining neck between the adjacent surfaces at high and low RH, respectively. Pull-off Force (nN) 40 Fig. 4: Pull-off force vs RH measured between a sharp SFM tip coated with OTS and a flat silicon sample. The pull-off force is independent of humidity. 30 20 10 0 0 20 40 60 80 100 Relative Humidity (%) Hence, based on the above results, the three regimes in pull-off force SFM measurements for adjacent hydrophilic surfaces (Fig. 3)can be interpreted as follows: In regime I, no capillary neck is developed, and the pull-off force is dominated by van der Waals interactions. A capillary neck is formed at about 40% RH, which corresponds to the force discontinuity observed between regimes I and II. We can understand this transition-like behavior of the pull-off force by considering the minimum thickness requirement of water precursor films for spreading(28,29). The height of the precursor film can not drop below a certain minimum, e, which is 1/ 2 A ; S SO SL . e a0 ; a0 (2) S 6 where a0 is a molecular length,(29), S the spreading coefficient, A the Hamaker constant, SO the solid-vacuum interfacial energy, and SL the solid-liquid interfacial energy. We propose that the formation of the capillary neck also requires a minimum height of the water film. No capillary neck forms between two surfaces until the water film thickness reaches the minimum thickness. The water film thickness was found to increase with the increase of RH (i.e., p/ps).(19), i.e., the thickness of the water film on the silicon surface is too thin to form a capillary neck with the probing tip for RH less than 40%. When the water film thickness reaches the minimum thickness requirement at 40% RH, a 1/ 2 6 2.1 Intermolecular and Surface Forces ChemE 554/ove capillary neck forms between the tip and the substrate surfaces, leading to a sudden increase of the pull-off force. The magnitude of pull-off forces measured on hydrophilic silicon surfaces below 40% RH is 83 nN (Fig. 3 and 4). For RH larger than the critical RH, in the mid-RH regime II (Fig. 2 and 3), the capillary force dominates the pull-off force if both surfaces are hydrophilic. Thus, the SFM observable the pull-off force is not a direct measure of the capillary force only. In regime II the pull-off force can be described as the sum of the capillary force (Fcap) and van der Waals interaction force (Fvdw), i.e., Fpull Fcap Fvdw (3) In regime I, the pull-off force is restricted to van der Waals interaction between the cantilever tip and the sample surfaces. Both Fcap and Fvdw are attractive. In the high RH regime III (Fig. 2 and 3), the pull-off force decreases with increasing RH for a hydrophilic tip. Mate and Binggeli(5) discussed the decrease as the interplay between capillary forces and the forces related to the chemical bonding of the liquid in the gap. This leads to the following expression for the pull-off force: p G a a Fpull Fcap Fvdw Fchem ; Fchem (4) kT ln z v v p s where Fchem(5) is the force related to the chemical bonding with G the Gibbs free energy, a the area of the liquid film, v the molar volume, the chemical potential. Measurements with hydrophilic cantilever tips on ionic surfaces, such as calcium fluoride, CaF2, show a similar qualitative trend in the pull-off force at low RH as found above on silicon surfaces. At intermediate RH, the pull-off force collapses very rapidly with increasing RH. This can be explained by ion-diffusion from calcium fluoride surface into the water film, which has a strong affect on the material properties such as the surface tension. Roughness effects can explain why force values for presumable microcontacts (silica glass sphere) at low loads are significantly smaller than expected from Eq. (1). The roughness of the sphere is 10 nm rms determined from a 2nd-order flattened AFM image over 1 m2 area of the sphere surface. At low load, the sphere makes contact with multiple nanosized asperities. This leads to a significant decrease in the pull-off force in the van der Waals interaction regime compared to an atomically smooth sphere. The argument also holds in the capillary regime. The force instability measured with silica glass spheres is widened by the asperity size dispersion, and the magnitude of the pull-off force is determined by the number of asperities in contact. Halsey and Levine suggested that the adhesive force between two rough spheres was dependent on the total amount of the fluid present.(30) Capillary force equation for nano-contacts We derived the capillary force equation for nano-contacts from the sphere-plane approximation, found in reference(25), with the distinction that we did not require a large contact area, and thus, do not restrict our capillary force equation to large sphere radii, R (Fig. 2). Starting from the surface free energy of the system, W,(25) W s c ; s (d 2 2R 2 sin 2 ) ; d R(1 cos ) , (5) 7 2.1 Intermolecular and Surface Forces ChemE 554/ove where is the angle of MOP , s the wetted surface area, and c a constant. The capillary force can be introduced as dW d F R 2 2 sin (1 cos ) . (6) dD dD where D is the distance between the sphere and the plane. The differential term of the angle with D can be obtained by an isovolume consideration (dV/dD = 0) of a simplified meniscus volume (ABMQN), V, which equals the volume of the cylinder ABMN minus the volume of the spherical cap MNQ. The simplified meniscus volume is R 3 (7) V R 2 sin 2 ( D d ) (1 cos ) 2 (2 cos ) 3 Equation (7) leads to the following relationship: d tan (8) dD D 2 R(1 cos )1 d This equation is also applicable to small contacts. The capillary force is derived by substituting equation (8) into equation (6), i.e., (1 cos ) 2 F R cos , (9) D cos 1 d which yields a capillary force at contact (D = 0) (1 cos )2 Rd . (10) Fcap Fmax R cos cos Equations (1) and (10) differ by the geometrical factor (1 cos ) 2 (11) K 4 cos which is important for small asperity contacts, i.e., large angles of Fig. 7). Equation (1) can be applied with a 20% uncertainty for an angle of less than 70. Yang and co-workers observed large pull-off forces (i.e., 100-200 nN) on mica with typical hydrophilic cantilever tips,(14) which we propose to explain with a large K factor. Note that Eq. (10) is based on a very simplified cylindrically shaped geometry. More sophisticated geometries are found in the literature for macrocontacts or microcontacts,(5,20-23) and for nanocontacts.(31) 8 2.1 Intermolecular and Surface Forces ChemE 554/ove 2.1.3 Van der Waals Forces Point Interaction Van der Waals forces exist between atoms or molecules and can be divided into three groups: Dipole-dipole force: Molecules having permanent dipoles will interact by dipoledipole interaction. Dipole-induced dipole forces: The field of a permanent dipole induces a dipole in a non-polar atom or molecule. Dispersion forces: Due to charge fluctuations of the atoms there is an instantaneous displacement of the center of positive charge against the center of negative charge. Thus at a certain moment a dipole exists and induces a dipole in another atom. Therefore non-polar atoms (e.g. neon) or molecules attract each other. The attractive van der Waals force between the atoms is proportional to 1/r7, where r is the distance between the atoms. The empirical potential often used is the LennardJones (LJ) potential: 1 12 6 A C C 6 C2 . ( r ) 6 12 4 ; ; r r 4B r A r The potential is also referred to as the 6-12 potential because of its (1/r)6 and (1/r)12 distance, r, dependence of the attractive interaction and repulsive component, respectively. The empirical constant represents the characteristic energy of interaction between the molecules (the maximum energy of attraction between a pair of molecules). , a characteristic diameter of the molecule (also called the collision diameter), is the distance between two atoms (or molecules) for (r) = 0. The LJ potential is depicted below. Examples for the LJ parameters, and , are provided in Table 2. Lennard Jones (6-12) potential (empirical Van der Waals Potential between two atoms or nonpolar molecules). 9 2.1 Intermolecular and Surface Forces ChemE 554/ove Table 2: Substance (Å) /k H2 (light element) Ar (noble gas) 2.915 3.418 38.0 124 Polyatomic Substances Air N2 3.617 3.681 97.0 91.5 Hydrocarbons CH4 n-C6-H14 3.822 5.909 137 413 k = 1.380 10-16 erg molecule-1 K-1 (Boltzmann's constant) Macroscopic Body Interaction Above a few Ångstroms to hundreds of Ångstroms, van der Waals forces are significant, particularly between macroscopic bodies. The interaction between different geometries, such two planes, a sphere and a plane, or two crossed cylinders, can be calculated by integration. For example, the attractive force between a sphere and a plane is F(D) = -AR/6D2 where R is the radius, D the distance between the sphere and the plane. The interaction constant A, is called Hamaker constant, defined as A=2C where C is the attractive interaction strength (see LJ potential above) and ii = 1,2, is the number density of the molecules in the solid (1 or 2). The figure below and Table 3 provide non-retarded Van der Waals interaction free energies between bodies of different geometries that were calculated on the basis of pairwise additivity (Hamaker Summation Method). Source: Intermolecular & Surface Forces, J. Israelachvili, Academic Press. (Attractive potentials: w and W) 10 2.1 Intermolecular and Surface Forces ChemE 554/ove Table 2 Notice: The energy of interaction between flat surfaces is per unit area! One notices the significantly impacted distance dependences. While, Van der Waals atom-atom interactions are very short ranged (~1/r6), macroscopic Van der Waals interactions are long ranged (e.g., sphere-sphere: ~1/D). The Hamaker Constant The Hamaker constant, A=2Cprovides the means to determine the interaction parameter C from the Van der Waals pair potential, w(r)=-C/r6. Typical values for A and the number density of molecules in a solid are provided below. The Hamaker constant demands pairwise additivity. Table 3: Medium Hydrocarbon CCl4 H2O C [10-79 Jm6] 50 1500 140 m-3 3.3 0.6 3.3 A [10-19 J] 0.5 0.5 1.5 Derjaguin Approximation The Derjaguin Approximation relates the force law, F(D), between two curved surfaces to the interaction free energy per unit area, W(D), between two planar surfaces. This makes this approximation a very useful tool, since it is usually easier to derive the interaction energy for two planar surfaces rather than for curved surfaces. The Derjaguin Approximation reads as follows: F ( D )curved 2R*W '' ( D )planar , where D is the separation distance, and R*is the combined curvature of the two surfaces, i.e., 1/R*=1/R1+1/R2. The Derjaguin Approximation is valid not only for additive inverse power law potentials (such as Van der Waals interactions) but for any type of force law, whether attractive, repulsive or oscillatory, as long as the range of the interaction and the separation distance D is much smaller than the curvature. To illustrate the power of the Derjaguin Approximation we will provide a derivation based on the inverse power law potential. We start with the pair potential 11 2.1 Intermolecular and Surface Forces ChemE 554/ove w (r ) C , rn (1) with the interaction parameter C. The interaction distance, r, is provided in figure 1 by r z2 x 2 . (2) Fig. 1 Now we let a single molecule of the sphere at distance D interact with the planar surface in figure 1 which yields the following interaction potential: zD x 0 w( D ) 2C dz z xdx 2 x2 n/2 2C dz 2C ;n 3 n 2 (n 2) D z ( n 2 )( n 3 )D n3 (3) If we consider the differential volume dV=x2dz==(2R-z)zdz provided by a thin circular section of the sphere of area x2 and thickness dz, the net interaction energy is W( D ) 2 2C 2 ( n 2 )n 3 z 2 R z 0 ( 2 R z )zdz ( D z )n3 (4) based on equation (3), a molecule-plane distance of D+z, and unchanged number density in the sphere and the plane. For very small distances (i.e., D<<R) only small values of z contribute which simplifies the integral as follows 2 2C 2 2 Rzdz , W( D ) ( n 2 )n 3 z0 ( D z )n3 (5) and yields the following final solution for the interaction energy between curved surfaces W( D ) 4 2C 2 R ( n 2 )n 3( n 4 )( n 5 )D n5 (6) 12 2.1 Intermolecular and Surface Forces ChemE 554/ove Note that the radius, R, in figure 1 represents the combined radius, R*, in the case of two curved surfaces. In the case of Van der Waals forces, i.e, n=6, the interaction energy becomes W ( D )curved 2C 2 R 6D (curved surfaces) (7) The force for curved surface interaction will therefore be F ( D )curved W ( D ) 2C 2 R (curved surfaces) D 6D 2 (8) Analogous one finds for planar surface interactions that W '' ( D )planar C 2 (planar surfaces), 12D 2 (9) which leads to the relationship F ( D )curved 2R*W '' ( D )planar . (10) confirming the Derjaguin Approximation. Example: For two spheres in contact (D=interaction distance), the interaction energy W() can be replaced by two times the surface energy, , which yields a contact interaction force for curved surfaces of F(D) curved 4R * . Hamaker Constant based on the Lifshitz Theory The assumption of the additivity ignores the existence of multiple reflections. Multiple reflections occur when atom A induces a dipole in atom B. At the same moment the field of atom A polarizes also another atom C. The induced dipole of atom C, influences atom B. Therefore the field of atom A reaches atom B directly and via reflection from atom C. The Lifshitz theory has overcome the problem of additivity. It is a continuum theory which neglects the atomic structure. The input parameters are the dielectric constants, , and refractive indices, n. The Lifshitz theory is in qualitative agreement with the results deduced by simple pairwise integration. The Hamaker constant for two macroscopic phases 1 and 2 interacting across a medium 3 is approximated as: 2 2 2 2 3 1 3 2 3 3h e n1 n3 n2 n3 , (11) A kT 2 2 2 2 4 1 3 2 3 8 2 n 2 n 2 n 2 n 2 n1 n3 n2 n3 1 3 2 3 13 2.1 Intermolecular and Surface Forces ChemE 554/ove where e is the absorption frequency (e.g., for H2O: e = 3 x 1015 Hz). Table 11.2 (below) provides non-retarded Hamaker constants determined with the Lifshitz theory (eq. 11). Source: Intermolecular & Surface Forces, J. Israelachvili, Academic Press. Retardation Effects The van der Waals forces are effective from a distance of a few Angstroms to several hundreds of Angstroms. When two atoms are a large distance apart, the time for the electric field to return can be critical, i.e., comparable to the fluctuating period of the dipole itself. The dispersion can be considered to be retarded for distances more than 100 Å, i.e., the dispersion energy begins to decay faster than 1/r6 (~1/r7). For macroscopic bodies retardation effects are more important than for atom-atom interactions (see Fig. below). 14 2.1 Intermolecular and Surface Forces ChemE 554/ove Non-retarded vs. retarded regime of attractive Van der Waals interaction between two mica surfaces of radius R 1 cm in water. 2.1.4 Adhesion and Surface Energies The energy of adhesion (or just adhesion), W", i.e., the energy per unit area necessary to separate two bodies (1 and 2) in contact, defines the interfacial energy 12 as: W '' 2 12 ; 12 1 2 2 1 2 , where i (i= 1,2) represent the two surface energies. Assuming two planar surfaces in contact, the Van der Waals interaction energy per unit area is A W1 D (see above), 12D 2 which was obtained by pairwise summation of energies between all the atoms of medium 1 with medium 2. Neglected have been the summation of atom interactions within the same medium, which yields additional energy terms, i.e., A W2 const . 2 , 12Do consisting of a bulk cohesive energy term (assumed to be constant), and an energy term related to unsaturated "bonds" at the two surfaces in contact (i.e., D = Do). Notice, contact cannot be defined as D = 0 due to molecular repulsive forces. Do is called the "cutoff distance". Hence the total energy of two planar surfaces at a distance D Do apart is (neglecting the bulk cohesive energy) 2 A 1 1 A Do 1 2 . W W1 W2 12 Do 2 D 2 12Do 2 D A In contact (i.e., D=Do) W = 0. In the case of isolated surfaces, i.e., D = , W = 2 . 12Do 15 2.1 Intermolecular and Surface Forces ChemE 554/ove Thus, in order to separate the two surfaces one has to overcome the energy difference A W=W(Do)- W(D=)=2 , which corresponds to the adhesive energy per unit area 12Do of W''=212. Hence, the interfacial energy can expressed as function of the Hamaker A constant and the cutoff distance: 12 . 24Do2 Cutoff Distance The challenge is to determine Do, which unfortunately cannot be set equal to the collision diameter (i.e., the distance between atomic centers). Let us assume a planar solid consisting of atoms that are close-packed. Each surface atom (of diameter ) will have nine nearest neighbors (instead of 12 as in the bulk). When surface atoms come into contact with a second surface each atom will gain (12-9)w=3w=3C/6 in binding energy. Thus, the energy per unit area, S=2sin(60 deg) = 2√3/2, is 1 3w 3C 3C 2 2 12 8 ; 3 2 2 S 2 where reflects the bulk atom density for a close packed system. Introducing the 3C 2 3A A definition of the Hamaker constant, it follows 12 2 2 2 2 2 2 24 2.5 A For = 0.4 nm and 12 it follows Do = 0.16 nm. Do = 0.16 nm is a remarkable 24Do2 "universal constant" yielding values for surface energies that are in good agreement with experiments as shown in the Table below. Source: Intermolecular & Surface Forces, J. Israelachvili, Academic Press. 16 2.1 Intermolecular and Surface Forces ChemE 554/ove References (Capillary Forces) (1) E Barthel, XY Lin, JL Loubet: Adhesion energy measurements in the presence of adsorbed liquid using a rigid surface force apparatus. Journal Of Colloid and Interface Science 177 (1996) 401-06. (2) J Crassous, E Charlaix, JL Loubet: Nanoscale investigation of wetting dynamics with a surface force apparatus. Physical Review Letters 78 (1997) 2425-28. (3) LR Fisher, JN Israelachvili: Experimental Studies on the applicability of the Kelvin Equation to Highly Curved Concave Menisci. Journal of Colloid and Interface Science 80 (1981) 528-41. (4) LR Fisher, JN Israelachvili: Direct measurement of the effect of meniscus forces on adhesion: a study of the applicability of macroscopic thermodynamics to microscopic liquid interfaces. Colloids and Surfaces 3 (1981) 303-19. (5) M Binggeli, CM Mate: Influence of capillary condensation of water on nanotribology studied by force microscopy. Appl. Phys. Lett. 65 (1994) 415-17. (6) L Xu, A Lio, J Hu, DF Ogletree, M Salmeron: Wetting and capillary phenomenon of water on mica. J. Phys. Chem. B 102 (1998) 540-48. (7) J Hu, X-D Xiao, DF Ogletree, M Salmeron: Imaging the condensation and Evaporation of molecularly thin films of water with nanometer resolution. Science 268 (1995) 267-69. (8) J Hu, X-D Xiao, M Salmeron: Scanning polarization force microscopy: a technique for imaging liquids and weakly adsorbed layers. Appl. Phys. Lett. 67 (1995) 476-78. (9) PB Miranda, L Xu, YR Shen, M Salmeron: Icelike water monolayer adsorbed on mica at room temperature. Phys. Rev. Lett. 81 (1998) 5876-79. (10) T Thundat, X-Y Zheng, GY Chen, RJ Warmack: Role of relative humidity in atomic force microscopy imaging. Surf. Sci. Lett. 294 (1993) L939-L43. (11) T Thundat, RJ Warmack, DP Allison, LA Bottomley, AJ Lourenco, TL Ferrell: Atomic force microscopy of deoxyribonucleic acid strands adsorbed on mica: the effect of humidity on apparent width and image contrast. J. Vac. Sci. Technol. A 10 (1992) 630-35. (12) M Fujihira, D Aoki, Y Okabe, H Takano, H Hokari: Effect of capillary force on friction force microscopy: a scanning hydrophilicity microscope. Chemistry Letters 7 (1996) 499-500. (13) M Odelius, M Bernasconi, M Parrinello: Two dimemsional ice adsorbed on mica surface. Phys. Rev. Lett. 78 (1997) 2855-58. (14) GL Yang, JP Vesenka, CJ Bustamante: Effects of tip-sample forces and humidity on the imaging of DNA with a scanning force microscope. Scanning 18 (1996) 344-50. (15) Y Sugawara, M Ohta, T Konishi, S Morita, M Suruki, Y Enomoto: Effects of humidity and tip radius on the adhesive force measured with atomic force microscopy. Wear 168 (1993) 13-16. (16) AL Weisenhorn, PK Hansma, TR Albrecht, CF Quate: Forces in atomic force microscopy in air and water. Appl. Phys. Lett. 54 (1989) 2651-53. (17) BV Derjaguin, NV Churaev: Structural component of disjoining pressure. J. Coll. Interf. Sci. 49 (1974) 249-55. (18) RM Pashley: Multilayer adsorption of water on silica: an analysis of experimental results. J. Coll. Interf. Sci. 78 (1980) 246-48. (19) D Beaglehole, HK Christenson: Vapor adsorption on mica and silica: entropy effects, layering, and surface forces. J. Phys. Chem. 96 (1992) 3395-403. (20) WC Clark, JM Haynes, G Mason: Liquid bridges between a sphere and a plane. Chem. Eng. Sci. 23 (1968) 810-12. (21) FM Orr, LE Scriven, AP Rivas: Pendular rings between solids: meniscus properties and capillary force. J. Fluid Mech. 67 (1975) 723-42. 17 2.1 Intermolecular and Surface Forces ChemE 554/ove (22) R Aveyard, JH Clint, D Nees: Theory for the determination of line tension from capillary condensation. Journal Of the Chemical Society-Faraday Transactions 93 (1997) 4409-11. (23) AW Adamson: Physical Chemistry of Surfaces, John Wiley & Sons, Inc., New York, 1990. (24) EA Vogler: Structure and reactivity of water at biomaterial surfaces. Adv. Coll. Int. Sci. 74 (1998) 69117. (25) JN Israelachvili: Intermolecular and surface forces, Academic Press, London, 1992. (26) G Meyer, NM Amer: Appl. Phys. Lett. 56 (1990) 2100. (27) R Luginbühl, A Szuchmacher, MD Garrison, JB Lhoest, RM Overney, BD Ratner: Comprehensive Surface Analysis of Hydrophobically Functionalized SFM Tips. Ultramicroscopy 82 (2000) 171-79. (28) R Bruinsma: Slow spreading of polymer melts. Macromolecules 23 (1990) 276-80. (29) PG de Gennes: Wetting: statistics and dynamics. Rev. Mod. Phys. 57 (1985) 827-63. (30) T Halsey, A Levine: How sandcastles fall. Physical Review Letters 80 (1998) 3141-44. (31) A Marmur: Tip-surface capillary interactions. Langmuir 9 (1993) 1922-26. 18 2.1 Intermolecular and Surface Forces ChemE 554/ove Adhesion between Rigid Spheres: Bradley Model Based on the discussion above (Derjaguin), the adhesion force between two rigid spheres can be expressed as Fadh 2R* ; 1 2 12 where is called the "work of adhesion" per unit area. This is the well-known Bradley model of adhesion. If compared with the elastic model by Johnston et al. (JKR Theory discussed in the Chapter Contact Mechanics), 3 JKR Fadh R* 2 which considers only adhesion over the contact area but an elastic response of the spheres, it seems as there is an inconsistency. as the JKR adhesion force is independent of any elastic property. We will resolve this issue in the following paragraph. Adhesion of elastic spheres: Bradley vs. Johnson (JKR), Tabor coefficient Adhesion of Elastic Spheres: The Tabor Coefficient 2.1.5 Varia Instrumentation: Surface Forces Apparatus, Optical Tweezers DLVO Theory 19 2.1 Intermolecular and Surface Forces ChemE 554/ove 20 2.1 Intermolecular and Surface Forces ChemE 554/ove 21