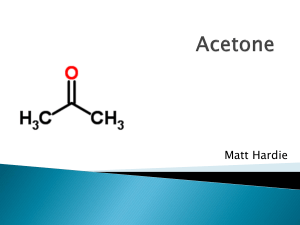
Oxygenates-Nature - Atmospheric Chemistry Modeling Group
advertisement

Dominant presence of oxygenated organic species in the remote southern Pacific troposphere H. Singh*, Y. Chen*, A Staudt#, D. Jacob#, D. Blake‡, B. Heikes**, J. Snow** *NASA Ames Research Center, Moffett Field, CA 94035; #Harvard University, Cambridge, MA 02138, ‡University of California, Irvine, CA 92697, **University of Rhode Island, Narragansett, RI 02882 _________________________________________________________________________ Oxygenated organic species are intimately involved with the fate of nitrogen oxides (NOx) and hydrogen oxides (HOx), which are necessary for tropospheric ozone formation1-2. A recent airborne experiment (March-April, 1999) focused over the southern hemisphere (SH) Pacific Ocean (PEM-tropics-B) provided a first opportunity for a detailed characterization of the oxygenated organic composition of the remote southern hemisphere troposphere. Three co-located multi-channel airborne instruments measured a dozen key oxygenated species (carbonyls, alcohols, organic nitrates, organic pernitrates, peroxides) along with a comprehensive suite of C2-C8 Nonmethane hydrocarbons (NMHC). These measurements reveal that in the tropical SH (0-30˚S), oxygenated chemical abundances are extremely large and collectively are nearly five times those of NMHC. Even in the NH remote atmospheres their burden is equal to or greater than that of NMHC. The relatively uniform global distribution oxygenates (Ox-org) is indicative of the presence of large natural and distributed sources. A global 3-D model, reflecting the present state of knowledge, is unable to correctly simulate the atmospheric distribution and variability of several of these species. Oxygenated organic species have the potential to play an important role in processes of atmospheric ozone formation1-5. Unlike most hydrocarbons, these species photolyze at tropospheric wavelengths and provide a ubiquitous source of atmospheric free radicals6-10. Multifunctional oxygenates may also form a substantial part of the organic component of global aerosol11-12. Measurements of these species in remote atmospheres are extremely sparse and virtually nonexistent in the SH. NASA/PEMtropics-B, an airborne experiment that utilized DC-8 (and P-3B) aircraft, provided a first opportunity for a detailed examination of the composition of the organic oxygenates in the SH remote troposphere. A dozen key oxygenated species and a comprehensive suite of some 28 C2-C8 NMHC were measured. This required the co-location and successful operation of three separate multi-channel airborne instruments. In addition, many other useful chemical and meteorological parameters were measured. Brief descriptions of these instruments, employing highly sensitive separation and detection techniques, and an overview of the experiment is being published separately13. A series of 18 DC-8 flights over the Pacific Ocean from 35˚N to 35˚S latitude over 90˚W to 150˚E longitude were performed. Nearly two thirds of these flights were in the SH. Extensive trajectory analysis and tracer measurements showed that direct impact of continental pollution was minimal. Thus the air sampled should represent near background conditions for this season. Figure 1 shows the mean vertical distribution of the most abundant oxygenated organic species measured during PEM-tropics-B. The top box (a) bins all the data collected in the SH (0-30˚S) and the bottom box (b) does the same for the NH (0-30˚N). The group containing reactive nitrogen is presented as total alkyl nitrates (TAN, RONO2) and peroxyacetyl nitrate (PAN, CH3C(O)OONO2). TAN is made up of several chemicals such as methyl nitrate, ethyl nitrate, i-propyl nitrate and n-butyl nitrate which were individually measured. TAN mixing ratios in the free troposphere were ≈10 ppt (10-12 v/v) and were as much as 60 ppt in the tropical marine boundary layer (MBL), because of a large marine source of methyl nitrate. Other alkyl nitrates (e. g. i-propyl nitrate) can have a near exclusive anthropogenic source14-15. PAN has no known primary sources and is produced as a result of atmospheric photochemical reactions between carbonyls (such as acetaldehyde, acetone) and NOx16. Free tropospheric (4-12 km) PAN mixing ratios in the NH (60-80 ppt) were substantially larger than those in the SH (20-30 ppt) mostly due to the greater availability of precursors. PPN (C2H5C(O)OONO2) was also measured but its mixing ratios were extremely small (<5 ppt) in comparison with PAN. Due to its fast thermal decomposition at warm temperatures, PAN declines rapidly towards the lower troposphere16. Formaldehyde (CH2O), acetaldehyde, and acetone were among the three carbonyls that were measured. Mixing ratios of CH2O (70-300 ppt) were nearly identical in the north and south tropical regions while those of acetaldehyde and acetone were somewhat lower in the SH. The unexpectedly high mixing ratios of acetaldehyde (60- 100 ppt) have been reported for the first time. Error bars in this measurement are of the order of a factor of two and presently this should be considered as an upper limit. Similarly, methanol concentration (≈900 ppt) was nearly two times that of ethane, the most abundant NMHC in the SH. In comparison to methanol, ethanol concentrations were rather low (<50 ppt). Methyl hydroperoxide was ubiquitous and one of the most abundant components in the SH lower troposphere (≈1000 ppt). Figure 2 shows the vertical distribution of the total abundance of oxygenated species (Ox-org) compared to total nonmethane hydrocarbons (C2-C8 NMHC). The NMHC included alkanes, alkynes, alkenes, and aromatics. Although heavier hydrocarbons (>C8) are likely present in the remote troposphere, the C2-C8 fraction of NMHC should capture >80% of the total NMHC burden17. At all altitudes in the SH troposphere (Figure 2a), Ox-org was nearly five times as abundant as C2-C8 NMHC. North of the equator (Figure 2b) Ox-org was about twice as abundant. These dramatic abundances have been observed for the very first time. Figure 3 shows the distribution of Ox-org and C2-C8 NMHC as a function of latitude for three altitude bins selected to represent the lower, middle, and upper troposphere. The rapid decline in the mixing ratio of C2-C8 NMHC from the NH to SH is evident. The same is not true for Ox-org as the mixing ratios in the SH are only slightly lower than the NH. Trajectory and tracer analysis indicated that the peak in the lower troposphere near 18˚N (Figures 2b and 3a) was attributable to pollution transport from the North American continent. The NH sources of NMHC are well known and their declining concentrations in the SH have been previously characterized17. No comparable data for the oxygenated species have been previously available. Figure 4 compares the mixing ratios of PAN, CH2O, acetaldehyde, and acetone observed over the South Pacific during PEM-Tropics (B) with results from the Harvard global 3-D model of tropospheric chemistry. This version of the model includes 80 chemical species (24 of them transported) to represent O3-NOx-hydrocarbon chemistry and is driven by assimilated meteorological winds from Florida State University18. Photochemically generated molecules such as PAN, CH2O and CH3OOH (not shown) are reasonably well simulated by this model although disagreements of the order of ±30% are present. The simulated CH3CHO concentrations, which over the South Pacific are mainly from the oxidation of ethane, are 10 times lower than observed. While methane photochemistry provides a large natural source of CH2O6,19, no viable alternatives for CH3CHO are currently known. We calculate that CH3CHO has a short atmospheric lifetime of about 1 day due to removal by reaction with OH and photolysis. The measured atmospheric burden and the associated error bars can be used to estimate a large global source 60-120 Tg y-1. Direct biogenic emissions of acetaldehyde are expected to be quite large but remain poorly quantified20-21. Acetaldehyde is known to be a common secondary product in the OH/O3 oxidation of nearly all NMHC4,22. This is also the case with acetone where terrestrial and photochemical sources of about 60 Tg y-1 have been identified23. The Harvard model under predicts the abundance of acetaldehyde and acetone in the southern Pacific and does not reproduce the low relative variability in the observed concentrations. These model underestimates can not be fixed by merely adding a primary source over the continents. It appears that a highly diffuse secondary production would need to be invoked in order to reproduce the observations. Lack of reliable source information on methanol prevented its simulation by the Harvard model. Like formaldehyde, methyl hydroperoxide also finds a major source in CH4/NMHC oxidation chemistry19. Methyl hydroperoxide can be highly effective in the transfer of HOx radicals from the lower to the upper troposphere8,10. The extremely high mixing ratio of methanol (≈900 ppt) in the SH troposphere is surprising. Although large terrestrial sources of methanol exist23, the possibility that oceans are strongly involved in its processing is indicated but todate has not been investigated. Strong seasonal cycles are expected to be present but their nature, reflecting a balance between sources and sinks, is not known at this time. Significantly elevated concentrations of methanol (5-20 ppb) and acetone (2-8 ppb) over the rural/forested areas have been reported in recent years20,24,25. Sources of acetaldehyde, acetone, and methanol alone (≥ 200 Tg y-1), are estimated to be more than double the total fossil fuel emissions of NMHC (≈100 Tg y-1)17. The near-uniform global Ox-org distribution implies the presence of large diffuse secondary sources associated with chemical reactions of hydrocarbons in the atmosphere. Oceanic emissions may also play an important role but to date have not been explored. In many instances, the oxidation products of NMHC (anthropogenic and biogenic) are more stable than the parent hydrocarbon and can exist in the atmosphere for a longer time period. In one model study26, the oxidation of pentane, a highly reactive HC, was studied in detail. While nearly 97% of pentane was oxidized within a period of 5 days, 85% of the organic carbon was still present when all the complex oxygenates were included. Recently it has been found that some 10-12% of highly reactive -pinene emissions rapidly produce acetone27. The carbon budget of several organic molecules (e. g. terpenes, aromatics) is poorly accounted for28 and keeps open the possibility of significantly large sources of unknown oxygenates. Using an exploratory proton transfer mass spectrometeric technique, Crutzen et al.29 reported detection of a variety of oxygenated chemicals that could not be easily explained from known tropical forest emissions. It is likely that many as yet undiscovered oxygenated species are present in the atmosphere. Most oxygenated organics impact atmospheric photochemistry by providing free radicals and sequestering NOx from the system generally in the form of PAN1. Acetone and acetaldehyde are particular examples of this. Except for the alcohols, the oxygenates discussed here photolyze at tropospheric wavelengths and are a direct source of free radicals. In general formaldehyde is an intermediate product of this active chemistry. The degree of HOx amplification varies from molecule to molecule depending on their dominant loss process. For example, a molecule of acetone when photolyzed can produce 3 molecules of HOx. Since acetaldehyde is largely depleted via reaction with OH, it can only amplify HOx by about 1.5. These oxygenated chemicals can allow the biosphere to exert direct control over the oxidative capacity of the atmosphere while not requiring ozone. Most of the oxygenates measured in this study are only moderately soluble in water. However, their solubility rapidly increases in acidic solutions and at very high concentrations of acids (e. g. 70% H2SO4) molecules such as CH3OH may produce reactive species such as CH2O in liquid phases. Li et al.12 find that acetaldehyde and acetone undergo condensation reactions on particles surfaces to form higher molecular weight carbonyl compounds. Thus it is likely that some of oxygenated species measured, along with the multifunctional but to date unmeasured species, can contribute significantly to the organic component of global aerosol. References: 1. Singh, H. B., M. Kanakidou, P. Crutzen, and D. Jacob, Nature 378, 50-54 (1995). 2. Wennberg P. O. et al., Science 279, 49-53 (1998). 3. NRC, Formaldehyde and other aldehydes, Nat. Acad. Press., Wash. DC, 1981. 4. Carlier, P., H. Hannachi, and G. Mouvier, Atmos. Environ. 20, 2079-2099 (1986). 5. Folkin, I. and R. Chatfield, J. Geophys. Res. 105, 11,585-11,599 (2000). 6. Levy, H. II, Science 173, 141-143 (1971). 7. Chatfield, R. B. and P. J. Crutzen, J. Geophys. Res. 89, 7111-7132 (1984). 8. Prather, M. and D. Jacob, Geophys. Res. Lett. 24, 3189-3192 (1997). 9. Müller, J. F. and G. Brasseur, J. Geophys. Res. 104, 1705-1715 (1999). 10. Jaeglé, L., D. Jacob, W. Brune, and P. Wennberg, Atmos. Environ., in press (2000). 11. Murphy, D. M., D. S. Thomson, and M. J. Mahoney, Science, 282, 1664-1669 (1998). 12. Li, P. et al., J. Geophys. Res. submitted (2000). 13. Raper, J. L. et al., J. Geophys. Res., submitted (2000). 14. Atlas, E. et al., J. Geophys. Res. 97, 10,331-10,348 (1992). 15. Fischer, R. G., J. Kastler, and K. Ballschmitter, J. Geophys. Res. 105, 14,473-14,494 (2000). 16. Singh, H. B., and Hanst, P. L., Geophys. Res. Lett. 8, 941-944 (1981). 17. Singh, H. B. and Zimmerman, P. B., Adv. in Env. Sci. and Technol. 24, John Wiley and Sons, New York, 177-235 (1992). 18. Bey, I. et al., J. Geophys. Res. submitted (2000). 19. Logan, J. et al., J. Geophys. Res. 86, 7210-7254 (1981). 20. Riemer, D., et al., J. Geophys. Res. 103, 28,111-28,128 (1998). 21. Kesselmeier, J. and M. Staudt, J. of Atmos. Chem 33, 23-38 (1999). 22. Atkinson, R. et al., Atmos. Environ 34, 2063-2101 (2000). 23. Singh, H. B. et al., J. Geophys. Res. 105, 3795-3805 (2000). 24. Goldan, P. D., Kuster, W. C., Fehsenfeld, F. C. and Montzka, S. A., J. Geophys. Res. 100, 25,945-25,963 (1995). 25. Lamanna, M. S., and A. H. Goldstein, J. Geophys. Res. 104, 21,247 -21,262 (1999). 26. Madronich, S. and Calvert, J. G., J. Geophys. Res. 95, 5697-5715 (1990). 27. Reissell, A., C. Harry, S. Aschmann, R. Atkinson, and J. Arey, J. Geophys. Res. 104, 13,869-13,879 (1999). 28. Seinfeld, J. and S. Pandis, Atmospheric chemistry and physics, John Wiley & Sons, New York, 1998. 29. Crutzen, P. J. et al., Atmos. Environ 34, 1161-1165 (2000). Acknowledgment: This research is supported by the NASA Global Tropospheric Experiment. We thank all PEM-tropics (B) participants for their support. 12 PEM Tropics-B; 0-30 o o o S; 150 E-100 W (CH ) CO 3 2 10 ALTITUDE, km CH OOH 3 TAN 8 CH OH PAN 3 6 4 CH CHO 3 2 HCHO 0 1 10 100 1000 Concentration, ppt 12 PEM Tropics-B; 0-30 o o o N; 150 E-100 W (CH ) CO 3 2 10 CH OOH ALTITUDE, km 3 TAN 8 CH OH 3 6 PAN HCHO 4 2 CH CHO 3 0 1 10 100 1000 Concentration, ppt Figure 1: Mean vertical distribution of oxygenated organics in the southern Pacific (030˚S) and the northern Pacific (0-30˚N) troposphere. TAN (total alkyl nitrates) represents the sum of several minor organic nitrates. All data are collected over the Pacific Ocean during March-April, 1999 over a longitude of 90˚W to 150˚E. PEM Tropics-B; 0-30 12 o S ALTITUDE, km 10 NMHC 8 6 4 2 Oxygenates 0 0 500 1000 1500 PEM Tropics-B; 0-30 12 2500 3000 o N Oxygenates 10 ALTITUDE, km 2000 8 6 4 NMHC 2 0 0 500 1000 1500 2000 2500 3000 Concentration, ppt Figure 2: Mean vertical distribution of total oxygenated organics (Ox-org) and total NMHC (C2-C8 NMHC) in the southern Pacific (0-30˚S) and the northern Pacific (0-30˚N) troposphere. PEM Tropics-B (150ÞE-100ÞW) 5000 8-12 km 4-8 k m 0-4 k m 0-2 k m Ox-org (ppt) 4000 3000 2000 1000 0 -40 -30 -20 -10 0 10 20 30 40 0 10 20 30 40 2500 8-12 km 4-8 k m 0-4 k m 0-2 k m NMHC (ppt) 2000 1500 1000 500 0 -40 -30 ÞS -20 -10 Latitude ÞN Figure 3: Latitudinal distribution of total oxygenated organics (Ox-org) and total NMHC (C2-C8 NMHC). The data are binned into three altitude bands representing the lower (0-4 km), middle (4-8 km), and upper (8-12 km) troposphere over the Pacific Ocean. 12 10 10 8 8 ALTITUDE, km ALTITUDE, km 12 6 4 2 6 4 2 0 0 0 10 20 30 40 50 0 100 200 300 400 500 CH O, pp t 12 12 10 10 8 8 ALTITUDE, km ALTITUDE, km PAN, p pt 6 4 2 2 6 4 2 0 0 0 20 40 60 ACETAL DEHYDE, ppt 80 100 0 100 200 300 400 500 ACETONE, pp t Figure 4: A comparison of measured (solid line) and 3-D model predicted (dashed line) distributions of PAN, CH2O, acetaldehyde and acetone. The Pacific Ocean region of 0-30˚S latitude and 165˚E-100˚W longitude is selected for this comparison. SGG:245-5 August 18, 2000 Editor NATURE 968 National Press Building Washington, DC 20045 Dear Editor, Please find enclosed four copies of a manuscript titled "Dominant presence of oxygenated organic species in the remote southern Pacific troposphere" by H. B. Singh and co-workers. We would like you to consider this manuscript for publication in NATURE in the section "Letters to Nature". The manuscript is based on airborne measurements and theoretical studies of the global troposphere. A list of possible reviewers is attached. We look forward to your response. Sincerely yours, Hanwant B. Singh Earth Science Division Mail Stop 245-5 Phone: (650) 604-4769 Fax: (650) 604-3625 E-mail: hsingh@mail.arc.nasa.gov Enclosure POSSIBLE REVIEWERS (arranged alphabetically) Dr. Hajime Akimoto Institute for Global Change Research, Yokohama Campus 3175-25 Showa-machi, Kanazawa-ku Yokohama, Kanagawa 236-0001, JAPAN akimoto@frontier.esto.or.jp Dr. Gregory R. Carmichael Dept. of Chemical and Biochemical Engineering University of Iowa Iowa City, IA 52240, USA gcarmich@icaen.uiowa.edu Dr. Paul J. Crutzen Max-Planck Institute for Chemistry Postbox 3060 D-55020 Mainz, GERMANY air@mpch-mainz.mpg.de Dr. Allen Goldstein College of Natural Resources 330 Hilgard Hall # 3110 Univ. of California at Berkeley Berkeley, CA 94720, USA ahg@nature.berkeley.edu Dr. Maria Kanakidou Department of Chemistry University of Crete P.O.Box 1470 71409 Heraklion, GREECE mariak@chemistry.uch.gr Dr. Stuart Penkett University of East Anglia School of Environmental Sciences Norwich NR4 7TJ, U. K. m.penkett@uea.ac.uk